
Case Presentation
J Pediatri Endocrinol. 2018; 3(1): 1022.
The Curious Case of a Hyperpigmented Neonate: Familial Glucocorticoid Deficiency Presenting at Birth
Shirodkar DG*, Anandi SV and Bhattacharyya S
Department of Pediatrics, Manipal Hospital, Manipal University, India
*Corresponding author: Shirodkar DG, Department of Pediatrics, Manipal Hospital, Manipal University, HAL 2nd stage, Old airport road, Bangalore 560017, India
Received: April 09, 2018; Accepted: May 09, 2018; Published: May 16, 2018
Abstract
Background: Familial glucocorticoid deficiency is a rare autosomal recessive disorder characterized by isolated glucocorticoid deficiency usually in the presence of normal mineralocorticoid function.
Case Presentation: A late preterm girl born to non consanguineous couple presented at 15 days of age in view of congenital pnemonia with metabolic acidosis and refractory shock. There was history of early unexplained deaths in her siblings, both were hyperpigmented. Birth weight was 2.86 kg (10th percentile) and length was 47 cm (25th percentile). On examination the baby had generalised hyperpigmention. There was no evidence of organomegaly, atypical genitalia or dysmorphic features.
Investigations revealed normal hemogram, electrolytes and blood sugar. There was no acidosis. Serum cortisol level was very low (12 nmol/L), with significantly elevated serum ACTH levels (14080 pg/ml). 17 OH Progesterone levels were normal. Plasma renin activity was more than 500mIU/ml with normal serum aldosterone level. Metabolic screening was normal. She was diagnosed as probable familial glucocorticoid deficiency. The child was started on hydrocortisone at 10 mg/m2/d following which there was improvement in clinical parameters. Next generation sequencing of genes MC2R and MRAP showed a heterozygous mutation in MRAP gene. On followup there was a decrease in hyperpigmentation with normal developmental milestones and a fall in ACTH levels (1160 pg/ml).
Conclusion: This rare diagnosis needs to be considered when a neonate presents with hyperpigmentation, unexplained sibling death and refractory shock without atypical genitalia. Genetic analysis may not always reveal a pathogenic mutation in 40% of cases due to its genetic heterogeneity.
Keywords: Hyperpigmentation; Neonate; Adrenal insufficiency; Familial glucocorticoid deficiency
Introduction
Generalized hyperpigmentation with adrenal insufficiency in the neonatal period is unusual. We report a case of a neonate with generalised hyperpigmentation and refractory shock. Familial Glucocorticoid Deficiency (FGD) is a rare autosomal recessive condition. It is characterized by isolated glucocorticoid deficiency in the presence of normal plasma renin and aldosterone levels. Patients may present with hypoglycemic seizures, hyperpigmentation, recurrent infections, failure to thrive and coma, all of which can be attribute to low cortisol levels [1]. Hyperpigmentation is almost always observed at presentation of FGD patients. Cases of this condition have been reported in the white [2,3], black, Indian [4,5] and Middle Eastern [6] populations.
Case Presentation
A 15 day old baby girl, born late preterm at 36 weeks, to a non consanguineous couple of Maldivian ethnicity, was referred to our centre for evaluation of refractory shock and congenital pneumonia with metabolic acidosis. The refractory shock was treated in a hospital at Maldives with inotropes and hydrocortisone. There was family history of death in two of her siblings. The first child was a male who died at 8 hours of life (cause unknown) and the third child was a female who died at 7 years of age, who suffered from epilepsy and had developmental delay. Both of them were deeply hyperpigmented. The second child is a male and is alive and doing well. Our case was the youngest of them all. On examination this child was hyperpigmented similar to her other 2 siblings with unexplained deaths. Child was tachypneic with a blood pressure of 80/60 mm Hg. The baby weighed 2.86 kg (10th percentile) and length was 47 cm (25th percentile). There was no organomegaly, no dysmorphism with normal female genitalia. Laboratory investigations revealed a normal hemogram, electrolytes, plasma ammonia and blood sugar. The chest radiograph and sepsis screen did not show any abnormality. Neonatal metabolic screen (inclusive of congenital hypothyroidism, congenital adrenal hyperplasia, galactosemia, G6PD deficiency, biotinidase deficiency and phenylketonuria) was normal. In view of hyperpigmentation, refractory shock requiring hydrocortisone, and previous sibling deaths we performed investigations to detect etiology of adrenal insufficiency. The morning sample for serum cortisol revealed severe hypocortisolemia [12 nmol/l (70-634)] and plasma ACTH levels were very high [14080 pg/ml (6-48)]. Plasma renin level was high [>500 mIU/ml (4-89)] and plasma aldosterone concentration was normal [31.70 ng/dl (5-90)]. 17-hydroxy progesterone level was low normal [0.2ng/ml (0.1-9.40)] and DHEA-S was low [0.07 microgram/ml (0.4-2.50)]. Ultrasound abdomen and pelvis demonstrated mullerian structures. CT abdomen did not show adrenal mass, hemorrhage or calcification. Chromosomal analysis revealed a normal 46 XX karyotype. A clinical diagnosis of Familial Glucocorticoid Deficiency (FGD) was made and started on treatment with hydrocortisone (10 mg/m2 /day). Next generation sequencing of genes MC2R and MRAP showed heterozygous variant c.173T>C/p. Leu58Pro in MRAP gene, probably a variant of unknown significance. However further testing of other genes also responsible for this condition like NNT and GCCD 3 could not done in view of financial constraints faced by the family members.
On follow up, 8 months after diagnosis, the hyperpigmentation had markedly decreased. Her length was 64.5 cm (3rd centile) and her weight was 8 kg (50th to 97th centile). Her milestones were appropriate for chronological age. Plasma ACTH had decreased significantly (1140 pg/ml) and plasma renin activity had normalised to 6.92 ng/ml/ hr (2.35-37). However in view of still elevated ACTH levels the dose of hydrocortisone increased to 12 mg/m2/day. A written informed consent for publication was taken from her mother.
Discussion
Generalised hyperpigmentation at birth with history of unexplained deaths in siblings pursued us to consider adrenal insufficiency. Differential diagnosis of generalised hyperpigmentation with adrenal insufficiency in neonates includes congenital adrenal hyperplasia, X-linked congenital adrenal hypoplasia, familial glucocorticoid deficiency, Allgrove syndrome and acquired conditions such as adrenal hemorrhage causing adrenal crisis [7]. Presence of low serum cortisol with excessively elevated ACTH, normal aldosterone production, normal electrolytes, correction of the shock after giving hydrocortisone and the exclusion of other causes of adrenal failure suggested isolated glucocorticoid deficiency. Derangement of the renin-angiotensin system and mild salt-wasting at the time of diagnosis have been reported in some cases [8,9]. High plasma renin levels in the presence of normal electrolytes, as in our patient were attributed to volume depletion and infection (stress) at the time of diagnosis. However, plasma renin activity and aldosterone levels on follow-up were normal.
Our patient was deeply hyperpigmented at birth. This is due to the fetal corticotrophs producing excessive plasma ACTH in response to low cortisol which acts on melanocytes via melanocyte stimulating hormone receptors (melanocortin 1 receptor) to produce melanin synthesis before birth which fades once proper treatment is initiated, as is the case in our patient [1,8,10-14]. In some FGD patients the plasma ACTH levels are difficult to normalise and needs large doses of hydrocortisone and they remain hyperpigmented. In our patient normalisation of ACTH was difficult too, hence the dose of hydrocortisone was increased.
Patients with FGD may have low/undetectable levels of adrenal androgens probably secondary to reduced adrenocortical inner zone cell number [15]. In our patient, 17 hydroxy progesterone was on the lower limit of normal for age and gestation and DHEA-S levels were very low. Mutations for MC2R and MRAP gene were negative in our patient which accounts for 45% of cases of FGD. However the newer gene mutations implicated in the causation of FGD were not analysed due to non availability of the services in our country. Other gene mutations implicated in FGD includes GCCD3 (Glucocorticoid Deficiency 3 mapping to 8q11.2-q13.2), NNT (Nicotinamide Nucleotide Transhydrogenase) and TXNRD2 (Thioredoxin Reductase 2), Mini Chromosome Maintenance Deficient 4 (MCM4), Glutathione Peroxidase 1(GPX 1), Peroxiredoxin 3 (PRDX3) and Steroidogenic Acute Regulator Protein (STAR) [3,16]. Study done by Chan et al [3] revealed Whole Exome Sequencing (WES) was not able to detect all genetic mutations within intronic/regulatory regions and novel mutations in the gene(s) (Figure and Table).
![]()
Investigation
Values (At presentation)
Normal range
Values at 8 months (most recent visit)
Adreno Corticotropic Hormone (ACTH)
14080 pg/ml
6-48 pg/ml
1140 pg/ml
Plasma Renin Activity
>500 mIU/ml
2.8-39.9 mIU/ml
2.35-37 ng/ml/hr
6.92 ng/ml/hr
Serum Cortisol
12 nmol/L
140-690 nmol/l
-
17 Hydroxy Progesterone
0.2 ng/ml
0.13-1.06 ng/ml
0.02 ng/ml
Thyroid Stimulating Hormone (TSH)
4.4 mIU/ml
0.9-7.7 mIU/ml
3.460 mIU/ml
Serum Aldosterone
31.7 ng/dl
5-90 ng/dl
9.72 ng/dl
Table 1:
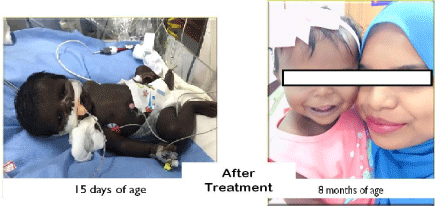
Figure 1:
Learning Points
1. This rare diagnosis needs to be considered when considering a case of hyperpigmentation, unexplained sibling death and refractory shock without atypical genitalia.
2. Early initiation of corticosteroid treatment leads to normalization of blood ACTH levels with improvement in clinical status and subsequent fading of the hyperpigmentation.
3. Early recognition and treatment may prevent mental disability and a fatal outcome, and identification of the causative mutation will enable future prenatal diagnosis in the family.
4. Genetic analysis will not always reveal a pathogenic mutation in more than 40% of cases due to its genetic heterogeneity [3].
References
- Ramachandran P, Penhoat A, Naville D, Begeot M, Osama Abdel-Wareth L, Reza SM. Familial glucocorticoid deficiency type 2 in two neonates. J Perinatol. 2003; 23: 62–66.
- Jazayeri O, Liu X, van Diemen CC, Bakker-van Waarde WM, Sikkema- Raddatz B, et al. A novel homozygous insertion and review of published mutations in the NNT gene causing Familial Glucocorticoid Deficiency (FGD). Eur J Med Genet. 2015; 58: 642–649.
- Chan LF, Campbell DC, Novoselova TV, Clark AJ, Metherell LA. Wholeexome sequencing in the differential diagnosis of primary adrenal insufficiency in children. Front Endocrinol (Lausanne). 2015; 6: 113.
- Vasudevan L, Joshi R, Das DK, Rao S, Sanghavi D, Babu S. Identification of novel mutations in STAR gene in patients with lipoid congenital adrenal hyperplasia: a first report from India. J Clin Res Pediatr Endocrinol. 2013; 5: 121–124.
- Jain V, Metherell LA, David A, Sharma R, Sharma PK, Clark AJ. Neonatal presentation of familial glucocorticoid deficiency resulting from a novel splice mutation in the melanocortin 2 receptor accessory protein. Eur J Endocrinol. 2011; 165: 987–991.
- Habeb AM, Hughes CR, Al-Arabi R, Al-Muhamadi A, Clark AJ, Metherell LA. Familial glucocorticoid deficiency: a diagnostic challenge during acute illness. Eur J Pediatr. 2013; 172: 1407–1410.
- Huebner A, Elias LL, Clark AJ. ACTH resistance syndromes. J Pediatr Endocrinol Metab. 1999; 12: 277–293.
- Clark AJ, Weber A. Adrenocorticotropin insensitivity syndromes. Endocrine Review. 1998; 19: 828–843.
- Davidai G, Kahana L, Hochberg Z. Glomerulosa failure in congenital adrenocortical unresponsiveness to ACTH. Clinical Endocrinology. 1984; 20: 515–520.
- Lu D, Haskell-Leuvano C, Inge Vage D, Cone RD. The melanocortin-1- receptor. In The Melanocortin Receptors, RD Cone. New Jersey: Humana Press. 2000; 309-339.
- Hunt G, Donatien PD, Lunec J, Todd C, Kyne S, Thody AJ. Cultured human melanocytes respond to MSH peptides and ACTH. Pigment Cell Research. 1994; 7: 217–221.
- Biller BMK, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. Journal of Clinical Endocrinology and Metabolism. 2008; 93: 2454–2462.
- Fluck CE, Martens JW, Conte FA, Miller WL. Clinical, genetic, and functional characterization of adrenocorticotropin receptor mutations using a novel receptor assay. Journal of Clinical Endocrinology and Metabolism. 2002; 87: 4318–4323.
- Weber A, Clark AJ. Mutations of the ACTH receptor gene are only one cause of familial glucocorticoid deficiency. Hum Mol Genet. 1994; 3: 585 –588.
- Weber A, Clark AJ, Perry LA, Honour JW, Savage MO. Diminished adrenal androgen secretion in familial glucocorticoid deficiency implicates a significant role for ACTH in the induction of adrenarche. Clin Endocrinol (Oxf). 1997; 46: 431–437.
- Meimaridou E, Hughes CR, Kowalczyk J, Guasti L, Chapple JP, King PJ. Familial glucocorticoid deficiency: new genes and mechanisms. Mol Cell Endocrinol. 2013; 371: 195–200.