
Case Presentation
J Pediatri Endocrinol. 2019; 4(1): 1026.
Albright’s Hereditary Osteodystrophy: a Rare Cause of Hip Avascular Necrosis: a Case Report
Salman A*
Department of Pediatric Endocrinology, Qassim University, Saudi Arabia
*Corresponding author: Almansour Salman, Department of Pediatric Endocrinology, Qassim University, Qassim, Saudi Arabia
Received: November 20, 2018; Accepted: January 28, 2019; Published: February 04, 2019
Abstract
Pseudohypoparathyroidism (PHP) is a rare genetic disease that is characterized by a clinical picture of hypoparathyroidism, but with a high level of Parathyroid Hormone (PTH) due to its resistance. Albright’s Hereditary Osteodystrophy (AHO) is a type of PHP that it has distinctive features such as obesity, short stature, brachydactyly and subcutaneous calcification. A wide variety of clinical and biochemical presentations have been reported in the past. We review here a case of AHO with rare presentation in pediatric populations. A four and half year old girl with a documented history of learning disability and attention deficit presented to the orthopedic clinic with the complaint of pain in the right hip, which was diagnosed as avascular necrosis of hip. During routine workup, she was found to have a low vitamin D level with high Thyroid Stimulating Hormone (TSH) and Parathyroid Hormone (PTH). She was managed initially as a case of Vitamin d dependent rickets with secondary hyperparathyroidism. The child was of comparatively short stature for her age, overweight, with a round face typical of AHO, dental caries and with short and broadened hands and feet. Her lab results showed normal calcium at 2.32 mmol/L, normal phosphate 1.78 mmol/L, high PTH 225 ng/L, high TSH 14.5 mU/L, normal Free-triiodothyroxine 17.8 P mol/L, and low Vitamin D3 (cholecalciferol) 40 nmol/L. An imaging study of her hand revealed short broad metacarpal and pharyngeal bones with low bone mass. Based on those clinical and lab findings, PHP was expected. The gene sequencing study of GNAS1 gene was sent and came positive for heterozygous mutation and confirmed the diagnosis of Albright’s Hereditary Osteodystrophy (AHO).
Keywords: Albright’s hereditary osteodystrophy; Hypoparathyroidism; Pseudohypoparathyroidism
Hereditary Osteodystrophy a Rare Cause of Hip Avascular Necrosis
Pseudohypoparathyroidism is a rare disease that was discovered initially by Fuller Albright and his colleagues in 1942 when they found group a of patients who presented with pictures of hypoparathyroidism (hypocalcemia, hyperphosphatemia) but their calcium and phosphate level failed to show responses after repeated injections of bovine parathyroid extract. It was concluded that the mechanism was end organ resistance that primarily impairs the renal actions of PTH, rather than deficiency in the hormones [1,2]. PHP was divided into type 1 and type 2, and type 1 subdivided to 1a, 1b, 1c, with the most common form being type 1a [3]. Presence of some distinctive features (short stature, rounded face, obesity, cutaneous ossification and metacarpophalangeal abnormalities) along with parathyroid hormone resistance indicate that which is known as Albright’s Hereditary Osteodystrophy (AHO) [4] [4].
PHP-1a is caused by heterozygous loss-of-function mutations in the Gs-alpha isoform of the GNAS gene on the maternal allele, which results in expression of the protein only from the paternal allele. Pseudopseudo hypoparathyroidism (PPHP) is a similar disorder characterized by features of AHO but with normal biochemistry and is caused by “mutations resulting in loss of function of the Gsalpha isoform of the GNAS gene on the paternal allele and resultant expression of the protein only from the maternal allele” [3,5,6]. Patients with type 1b have Renal PTH resistant hypocalcaemia and hyperphosphatemia, and imprinting/methylation defects at the GNAS locus, but they lack the features of AHO [7].
Many patients have been described with normocalcemia, with resistance to PTH [5]. Patients with PHP1a can present with hormonal resistance other than PTH such as TSH, Growth Hormone-Releasing Hormone (GHRH), gonadotropins and calcitonin. Hypothyroidism secondary to TSH resistance is common and has been the presenting feature in some neonates, and the explanation behind this is that the Thyroid-Stimulating Hormone (TSH) receptor is also coupled to adenylyl cyclase by Gs proteins, which is coded by the GNAS protein [1,3,6,8,9,10].
Case Presentation
Herein we report a case of pseudohypoparathyroidism type-1a in a child who presented with avascular necrosis of the hip. This etiology is not one that is typical or well documented, and that highlights the importance of this as a rare presentation.
A four and half year old girl presented to the orthopedic clinic in a local hospital with the complaint of pain in her right hip for the last year prior to presentation. The x-ray showed deformity and fragmentation of the right hip joint. The patient was referred to our hospital and her condition was diagnosed by our orthopedic surgeon as avascular necrosis of right hip. This child had been born preterm at 34 weeks, and was delivered by caesarian section with no problems in the neonatal period. She was referred to the endocrine clinic initially at age of 5 years because of Cushioned features. She has a history of learning disability and attention deficit, a medication history of using prednisolone only for several days (unknown dosage and duration), and a documented history of gaining weight and constipation.
There was no history of abnormal movement, with no vomiting, diarrhea, fractures or decrease in activity. She was on a regular diet with only occasional sun exposure, and her parents were consanguineous with no similar family history of complaints. Initially she was treated as a case of vitamin D-dependent rickets with secondary hyperparathyroidism (after showing low vitamin D level with normal ACTH and cortisol) and she was started on calcium Sandoz 500 mg once daily and cholecalciferol 6 drops daily.
Her vital signs were: heart rate 62 beats per minute, blood pressure 116/69 with normal temperature and oxygen saturation. Patient’s height was 96 cm (at-1.69 SDS), her weight was 20.2 kg (on 90th percentile), and her appearance was overweight with rounded face, dental caries, no striae, and no bone deformities. This patient was able to walk freely without limping, and there was no noted alopecia. The patient has short, broad hands and feet that will become more obvious later on life (Figure 1,2) with leg length discrepancy. Her lab results showed calcium 2.32 mmol/L (2.10-2.60), phosphate 1.78 mmol/L (1.20-1.95), PTH 225 ng/L (15-65), TSH 14.5 mU/L (0,27-4.20), Free triiodothyroxine 17.8 Pmol/L (12-22), alkaline phosphatase 290 U/L (100-240), Vitamin D3 (cholecalciferol) 40 nmol/L (61-200). The imaging-study of the patient’s hand revealed short broad metacarpal and pharyngeal bones with low bone mass (Figure 3).

Figure 1: Shortened first, fourth and fifth digit in both hands.
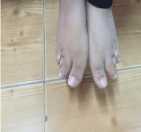
Figure 2: Brachydactyly of the feet, shortened third, fourth and fifth digits in
both feet were observed on physical examination.
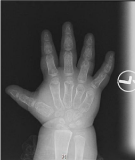
Figure 3: Short broad metacarpal and pharyngeal bones with low bone mass,
and advanced bone aging.

Figure 4: Yu, Campbell, Ciaffatelli et al 2014 [21].
Based on these clinical and lab findings, we expected to find Pseudohypoparathyroidism (PHP). The gene sequencing study of GNAS1 gene resulted in a heterozygous likely pathogenic. NM_000516.4: c.432+4A>G variant was detected and confirmed the diagnosis of AHO. During her follow up she was started on thyroxin 25 mcg then increased to 50 mcg as she was showing persistent high TSH.
Discussion
Examinations, testing, diagnostics and referrals for babies and children are very specialized fields of work, and unfortunately, many children and families with complex symptoms, syndromes and diseases are do not have access to specialized clinics or labs. Some do not live in areas where well-care is possible, and in rural and other locations, there may not be any hospital for many miles. Very often, and in healthcare settings that range from clinics to hospitals to private practices, it is very important that the first medical professional who examines the child be aware of signs and syndromes that indicate the presence of AHO or other diseases or conditions.
Lacking specific knowledge, training or adequate time for a full examination, there are a wide variety of problems and symptoms that may not be recognized as indicative of a more serious condition.
An example of such unintentional inaccuracies in children was noted in a study that which objective was to determine if there were one or more serious indicators for disease or syndromes in a group of children under the age of six years who were categorized as “obese”, and vice versa. A broad spectrum of testing was performed, including height/weight, Body Mass Index (BMI), presence/usage of narcotics, genetic history and other relevant factors. The critical focus of this study was whether all significant and relevant medical and other information were present in the respective health records for each child in a comparative Electronic Health Record (HER) search. There were a variety of undocumented diseases and conditions found in the testing and including AHO.
As noted in the case details and the lab reports, the child in this case study has notable dental caries but is without malformation of bone structure in the facial/oral regions. A study from India focused on the importance of dentists being aware of the sensitivity and fragility of teeth and bones of those who have parathyroid disorders. The authors note that approximately half of all patients (with thyroid conditions) have no symptoms or nonspecific symptoms, and that the thyroid conditions are found after further examination and testing. A wide array of typical complaints and problems are noted for these patients, including but not limited to pain, non-typical spacing of teeth and sensitivity [11]. They indicate that delayed recognition and treatment of these conditions can require orthopedic intervention and recommend that better awareness of thyroid and related conditions be part of routine and general dental care.
AHO was found to exhibit genetic features that did not always appear in patients at the same time-with some factors being obvious at birth or in childhood (dental anomalies, intellectual differences, structures of the hands and feet) and others not being noticed or diagnosed until adulthood (dental and cutaneous differences, osteo anomalies and the diagnosis of rickets, thought to be a result of difficulty in absorbing vitamin D). As noted in the case study presented here, diagnosis is sometimes contingent upon the patient’s/ family’s access to medical care that extends beyond routine well-care and emergency treatment. Patients and families whose skills or habits do not include documentation of symptoms, medical services or medicines may unwittingly elude accurate diagnosis and treatment for AHO and its related syndromes. This appears to be the situation of the child in the current case.
The study of AHO is ongoing, and the connections between AHO and PHP/PPHP demonstrate a variety of symptoms and responses to treatment. An earlier study examined the etiologies of PHP types, PPHP and also the connections between these syndromes and AHO. The authors noted that “AHO is difficult to diagnose because some clinical features are not obvious at birth or shortly after and may be very heterogeneous later. In case of PHP-Ia and PHP-Ib, laboratory analysis of endocrine parameters is mandatory but sometimes misleading…” [12]. This phenomenon may help to explain why the child in this current case study was not diagnosed at an earlier date. Some findings relevant to this case were discovered recently in a Hong Kong study, which notes that “It appears that PHP-1a is an autosomal dominant disease in which full clinical and metabolic abnormalities may not be present initially, but become apparent later” [13]. In that study, a mother and son (age 16) both demonstrated various symptoms that did not all present at early times in their respective lives.
It has been noted that AHO includes both a known array of features and those that appear unexpectedly, but with clear relationship to the primary disease. Among noted occurrences is that “Progressive Osseous Heteroplasia (POH) is a rare manifestation of AHO characterized by severe heterotopic ossification, which, unlike those typically observed in AHO patients, is progressive and affects deep connective tissue and skeletal muscle” (that) “can lead to ankylosis of affected joints and growth retardation of affected limbs later in life” [14]. Findings such as this, particularly when the skeletomuscular systems are involved, are clinical indications that AHO has physical and clinical ramifications that are still to be researched.
“Pseudohypoparathyroidism (PHP) is a heterogeneous group of rare endocrine disorders characterized by normal renal function and resistance to the action of Parathyroid Hormone (PTH), manifesting with hypocalcemia, hyperphosphatemia, and increased serum concentration of PTH. Genetic testing for a mutation in the GNAS1 gene can confirm diagnosis and identify subtype” [15]. Another study during that year focused on a 41 year old male, and noted that “The patient in this case presented with the classic and non-classic features of Albright’s hereditary osteodystrophy”, and found that, although the patient had a spectrum of symptoms that are not often associated with AHO, “these symptoms and associated pathologies are considered non-classic, much research has concluded that these presentations are quite commonly associated with pseudohypoparathyroidism” [3]. It has become quite clear that, despite very specific physical, skeletal and cutaneous presentations, along with an array of genetic and lab reports, there are inconsistencies in how AHO may affect patients on an individual basis.
In regard to bone anomalies caused by AHO, one study has recognized the tendency for children with this disease to both experience early bone maturation and abnormal epiphyseal fusion, but also indicates that “the epiphyses of long bones may remain open, thus an increase in height with growth-hormone therapy is still possible” [16]. Of course, each patient’s treatment is contingent upon a spectrum of symptoms and testing results, but this finding does indicate hope for those whose stature can be increased. The study does note that use of growth hormone requires close monitoring due to potential complications with the metabolic system, and that some healthcare insurance programs may be reluctant to consider this. The researcher expresses concern that, without knowledge of the very specific presentations of AHO, a child may be diagnosed with a primary symptom of obesity.
Avascular necrosis can occur in children and it manifests in two types: idiopathic and traumatic types. Both of them can affect the child’s growth and internal mechanisms that lead to a number of symptoms and conditions that are still not fully understood [17,18]. Among the causes is Legg-Calvé-Perthes Disease (LCPD). “The disease has an insidious onset and may occur after an injury to the hip” [19] and is ruled out contingent upon lab reports and history. An earlier study found that “Perthes disease may reflect a wider vascular phenomenon that could have long-term implications for the vascular health of affected individuals” [18], which indicates that vascular dysfunction in children requires extensive testing to determine both the etiology and the prognosis, as other serious symptoms may develop.
Because of the number of cases of infection, cancer and other diseases and conditions that are undiagnosed when initially seen, a medical team has provided a case study of an infant, whose situation was more complicated than that of a child who has language and speech capacity. This infant exhibited signs of pain and unwillingness to bear weight on one leg. The affected leg and foot were tender and warmer than the other foot, and most of the labs were within normal range. This report discusses some of the differences between conditions that can mask as less serious infections, diseases or injuries, and among the recommendations is to obtain an MRI as soon as possible. The article uses the acronym “LIMPSS” [20] to identify the issues and steps involved in making accurate diagnoses for pediatric complaints.
It is noted in the case study that the patient was originally believed to exhibit rickets, due to low levels of vitamin D. A study of patients with rickets, which sometimes has similar features of AHO, determined that “AVN is an independent risk factor for acute care utilization in patients with SCD” [21-25]. These researchers caution that if AVN is allowed to advance in severity, increased hospitalizations will be required, and they thus urge early treatment and prevention. Their estimation of hospitalization rates for those (primarily children) with AVN is displayed below.
While their study was focused on children with rickets, as opposed to AHO, the incidence of AVN would indicate the importance of early diagnosis and treatment to prevent exacerbation of bone deterioration, loss of mobility and accompanying pain and distress.
Conclusion
As evidenced in the case presented here, as well as other referenced studies, there are certain conclusions that can be drawn for referrals and testing for patients with the appearance of rickets, PHP, PPHP, AHO and AVN whose etiologies and prognoses are not definite. When there is no history of hereditary involvement in the suspected disease or syndrome, it is wise to do some additional labs (such as PTH) in bone disease practices or in the orthopedic clinic. Even with normal calcium, abnormality in PTH cannot be ruled-out, and discovery may lead to additional factors that will assist in diagnosis and treatment. As documented in the referenced studies, above, symptoms such as obesity, dental and orthodontic complaints, mobility difficulties and variances in facial features and the structure of hands and feet can mislead even experienced medical professionals as to the etiology and disease. Particular care must be taken with infants and those who are not able to verbally express their discomfort or other distress due to cognitive impairment or age.
In addition to family practitioners, primary physicians, physician assistants and nurse practitioners, there is a need for nurses, dentists, orthodontists and other specialists to be familiar with the symptoms and the routes for follow-up examinations and testing for patients of all ages who exhibit signs of AHO and the related diseases and conditions. Awareness of potential increased severity of symptoms and the development of related medical issues can enable interventions that will preserve mobility, motor function, dental integrity and overall quality of life.
Acknowledgement
I would like to thank Dr.Abdullah Alashwal for his support in this case.
References
- Bastepe M, Juppner H. Pseudohypoparathyroidism and Mechanisms of Resistance toward Multiple Hormones: Molecular Evidence to Clinical Presentation. The Journal of Clinical Endocrinology & Metabolism. 2003; 88: 4055-4058.
- Maeda SS, Fortes EM, Oliveira UM, Borba VC, Lazaretiti CM. Hypoparathyroidism and Pseudohypoparathyroidism. Arq Bras Endocrinol Metab. 2006; 50: 664-673.
- Kuzel AR, Lodi MU, Rahim M. Classic and Non-Classic Features in Pseudohypoparathyroidism: Case Study and Brief Literature. Cureus. 2017; 9: e1878.
- Wilson LC, Trembath RC. Albright’s hereditary osteodystrophy. J Med Genet. 1994; 31: 779-784.
- Yasuko T, Seiji K, Hiroko S, Toshihiro T, Toshimasa A. Pseudohypoparathyrodism Type 1a Patient with Normocalcemia. Endocrine Journal. 2008; 55: 169-173.
- Davies SJ, Hughes HE. Imprinting in Albright’s hereditary Osteodystrophy. J Med Genet. 1993; 30: 101-103.
- Kinoshita K, Minagawa M, Anzai M, Sato Y, Kazukawa I, Shimohashi K, et al. Characteristic Height Growth Pattern in Patients with Pseudohypoparathyroidism: Comparison between Type 1a and Type 1b. Clin Pediatr Endocrinol. 2007; 16: 31-36.
- Anne-Sophie B, Miriam L, Fritz-Line V, Virginie V, Catherine C, Michèle d, et al. Hypothyroidism in patients with pseudohypoparathyroidism type Ia: clinical evidence of resistance to TSH and TRH. Eur J Endocrinol. 2008; 159: 431- 437.
- Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001; 22: 675-705.
- Virginie VG, Michele D, Pascal P, Armelle F, Catherine B, Marc D, et al. Pseudohypoparathyroidism Ia and Hypercalcitoninemi. The Journal of Clinical Endocrinology & Metabolism. 2001; 86: 3091-3096.
- Mittal S, Gupta D, Sekhri S, Goyal S. Oral manifestations of parathyroid disorders and its dental management. J Dent Allied Sci. 2014; 3: 34-38.
- Montovani G. “Pseudohypoparathyroidism: Diagnosis and Treatment.” Journal of Clinical Endocrinology and Metabalism. 2011; 96: 3020-3030.
- Tam VH, Sammy PC, Chloe MM, Fung LM, Lee CY, Albert YC. “A novel mutation in pseudo hypoparathyroidism type 1a in a Chinese woman and her son with hypocalcaemia.” Hong Kong Medical Journal. 2014; 20: 258-260.
- Turan S, Murat B. “GNAS Spectrum of Disorders.” Current Osteoporosis Reports. 2015; 13: 146-158.
- Abraham, Mini R, George TG. “Pseudo hypoparathyroidism: Practice Essentials, Pathophysiology, Etiology.” Diseases & Conditions - Medscape Reference. Last modified. 2017.
- Nwosu BU. Pseudohypoparathyroidism in Children. Contemporary Aspects of Endocrinology. 2011; 433-442.
- McCarthy RE. Avascular necrosis of the femoral head in children. Instr Course Lect. 1988; 37: 59-65.
- Perry DC, DJ Green, CE Bruce, D Pope, P Dangerfield, MJ Platt, et al. “Abnormalities of Vascular Structure and Function in Children with Perthes Disease.” Pediatrics. 2012; 130: e126-e131.
- Harris GD, William LJ. Legg-Calve-Perthes. Disease: Background, Pathophysiology, Etiology. Diseases & Conditions-Medscape. 2018.
- Raam R, Jhun P, Bright A, Herbert M. Limping Child? Think LIMPSS. Annals of Emergency Medicine. 2016; 67: 297-300.
- Tiffany Y, Timothy C, Ciuffetelli I, Carlton H, Christopher PC, Linda R, et al. “Symptomatic Avascular Necrosis: An Understudied Risk Factor for Acute Care Utilization by Patients with SCD.” Southern Medical Journal. 2016; 109: 519-524.
- Daniel CP, Daniel JG, Colin EB, Daniel P, Peter D, Mary JP, et al. Abnormalities of Vascular Structure and Function in Children With Perthes Disease. AAP. 2012; 130.
- Brady CC, VV Thaker, T Lingren, JG Woo, SS Kennebeck, B Namjou-Khales, et al. “Suboptimal Clinical Documentation in Young Children with Severe Obesity at Tertiary Care Centers”. International Journal of Pediatrics. 2016; 4068582.
- Germain-Lee, Emily Germain-Lee. Albright Hereditary Osteodystrophy. 2015.
- Talal JA, Tan QYJ, Tong YG, Shenoy R, Kayani B, Parratt T, et al. “The role of electrical stimulation in the management of avascular necrosis of the femoral head in adults: a systematic review.” BMC Musculoskeletal Disorders. 2017; 18: 319.