
Research Article
J Pediatri Endocrinol. 2023; 8(1): 1057.
Gonadotropin-Releasing Hormone Studies to Assess Pubertal Deficiencies in Prader-Willi Syndrome
Melissa Kreutz1,2#; Ani Galfayan1,2#; Diane Stafford3*; Manaswitha Khare1,4#; Abhilasha Surampalli1; Brinda Patolia1; June-Anne Gold1,5; Suzanne B Cassidy6; Virginia Kimonis1,7#
1Division of Genetics and Genomic Medicine, Department of Pediatrics, University of California, USA
2Western University of Health Sciences, College of Osteopathic Medicine, USA
3Division of Pediatric Endocrinology, Department of Pediatrics, Stanford University School of Medicine, USA
4Division of Pediatric Hospital Medicine, Department of Pediatrics, Rady Children’s Hospital, USA
5Division of Medical Genetics, Department of Pediatrics, University of Loma Linda, USA
6Division of Medical Genetics, Department of Pediatrics, University of California San Francisco, USA
7Children’s Hospital of Orange County, USA
#These authors have contributed equally to this article.
*Corresponding author: Diane Stafford Division of Pediatric Endocrinology, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA, USA. Email: vkimonis@uci.edu; dejs@stanford.edu
Received: February 22, 2023 Accepted: March 29, 2023 Published: April 05, 2023
Abstract
Patients with Prader-Willi Syndrome (PWS) are known to have variable pubertal development with most having delayed or incomplete puberty. This study aims to evaluate the activity of the Hypothalamic-Pituitary-Gonadal (HPG) axis in individuals with PWS. Thirty-five patients (18 males, 17 females) ages ranging from 3 to 38 years with genetically verified PWS underwent GnRH stimulation testing using gonadorelin along with clinical evaluations. Of patients in the typical age range for initiation of puberty, six had LH peak >5IU/L, indicating activation, but the remaining six had variable peaks, indicating lack of, or indeterminate activation. In some females, physical examination noted breast development to Tanner stage II to III, despite apparent lack of HPG axis activation, while in some males testicular volumes were smaller than expected despite apparent activation of the HPG axis. We studied genotype differences (deletion vs UPD) with respect to HPG axis activation. Mean LH levels were nearly double for PWS females with UPD compared to PWS females with deletion. Both peak LH and peak FSH levels appear to be higher in males with deletion compared to UPD. Inconsistency between physical examination and stimulation testing results and the high variability in the age at activation of the HPG axis accentuates the need for the evaluation of pubertal staging in patients with PWS, and consideration of sex-steroid supplementation.
Keywords: Endocrine; LH; FSH; Deletion; Uniparental disomy
Introduction
Prader-Willi Syndrome (PWS) is a complex, multi-system syndrome that occurs at a frequency of approximately 1/10,000-1/30,000 and is equally distributed among males and females of all ethnic groups [1]. PWS results from loss of expression of paternally inherited genes on the chromosome 15(15q11.2-q13) by a variety of mechanisms which include large deletions (70-75%), maternal uniparental disomy for chromosome 15, UPD15 (20-30%), imprinting defects (2-5%), or rarely balanced translocations [1]. The deletion subtype of parental chromosome 15 is further divided into type I and type II deletions depending on chromosome breakpoint assignments [2]. Some of the patients with UPD 15 have both copies of the two maternal chromosomes 15 (heterodisomy: one copy each from the maternal grand-mother and one from the maternal grand-father), where as others inherit a duplicated single maternal 15(isodisomy). Duplication of deleterious genes in isodisomy could also cause differences between those patients and others with PWS and could point to candidate genes in the region. Clinical diagnosis in infancy and early childhood can be made on the basis of hypotonia with poor suckling in infancy, dysmorphic features including bitemporal narrowing, upslanting palpebral fissures, high palate, small chin, small hands and feet, and gonadal hypoplasia.
PWS is associated with multiple variable endocrine abnormalities, including hypogonadism, central hypothyroidism, Growth Hormone (GH)/insulin-like growth factor I axis dysfunction, and occasional central adrenal insufficiency. These are felt to be due to hypothalamic dysfunction and can result in short stature and delayed pubertal development. The most significant complications of PWS are related to uncontrollable hyperphagia due to hypothalamic dysfunction which results in severe obesity and increased risk of developing type 2 diabetes mellitus if food intake is not controlled externally. This has been reported in 25% of the adult PWS population [3,4]. Decreased lean body mass is common due to the combination of growth hormone deficiency and hypogonadism [1,5-7]. Obesity resulting from PWS in adult patients is distinguishable from exogenous obesity through the dominance of fat mass over lean body mass [6]. Active treatment with GH beginning prior to two years of age has been shown to improve body composition, motor function, cognition, height, and lipid profiles [5,7,8].
While there is a significant variability in the reproductive phenotype of those with PWS, there is commonly dysfunction of the HPG axis that is notable at birth [9]. Individuals with PWS often show clitoral and labia minora hypoplasia in females and a small penis with hypoplastic scrotal sac in males [7]. Also, unilateral or bilateral cryptorchidism is detected in 8090% of males [2]. Further, hypogonadism represents a common finding in adolescence leading to a range of outcomes from normal puberty to stalled puberty to complete or partial pubertal failure due to insufficient secretion of the pituitary gonadotropins LH and FSH and gonadal sex steroids. In adulthood, those with PWS are generally infertile with rare exceptions. Most women have either lack of or irregular menstruation and most men have either undetectable or low testosterone levels [10].
The aim of the current study is to determine the frequency of reproductive dysfunction based on GnRH agonist stimulation testing in a cohort of 35 individuals with PWS, comparing the incidence among the common subtypes, deletion vs UPD, and correlation with physical examination of pubertal status.
Materials and Methods
Subjects: Research protocols were approved by UC Irvine Institutional Review Board, and written informed assents and consents were obtained from all eligible participants or their legally responsible caregivers. Affected subjects aged 3 years to adulthood of both sexes were recruited into the study. Data was collected on 35 individuals with PWS who underwent GnRH stimulation testing to assess for pubertal development as part of a phenotype-genotype correlation study. Participants had a genetically and clinically confirmed diagnosis of PWS by methylation, and FISH and UPD analysis. Individuals who were FISH negative had UPD analysis if parents were available and those who had negative FISH and UPD analysis but were methylation abnormal were presumed to have imprinting center defects and eliminated from analysis because of the small sample size.
Thirty-five individuals with genetically verified PWS underwent GnRH stimulation testing using gonadorelin (2.5ug/kg. up to 100ug). GnRH stimulation testing involves giving a GnRH analog (gonadorelin or leuprolide) to stimulate the pituitary and then monitoring of LH and FSH levels occurred over time. Blood was drawn at baseline, 30 minutes, 60 minutes and 90 minutes post-gonadorelin administration through a standard protocol to measure serum LH and FSH levels at each time interval. If the HPG axis has been activated, there would be an increase in the LH and FSH levels after administration. Lack of rise implies absent activation of the HPG axis [10].
Physical examinations to gather data on breast Tanner staging or testicular volume in all 35 individuals were completed concurrently with the gonadorelin testing. Physical examinations were conducted uniformly by the Same Clinical geneticist (SC).
Data analysis: All data had a normal distribution, based on a p-value>0.05 with Levene’s test for normal distribution. The effect of genetic subtype was tested in pertinent variables of interest between groups using two-tailed, two-sample equal variance t-tests. Statistical significance was based on a p-value of <0.05 for all tests. The results were presented as mean +/-SD.
Results
Thirty-five patients underwent GnRH stimulation testing with gonadorelin. Of these, 18 were male and 17 were female (Table 1). A total of 16 had UPD and 19 had classic 15q deletions. The age range was slightly wider for the female patients with a maximum age of 38 years. All patients below 9 years of age had undetectable LH levels indicating lack of GnRH activation. An LH peak >5IU/L indicates activation of the HPG axis and onset of pubertal development. There was high variability in the group between 9 and 17 years, indicating lack of pubertal development in some patients, and activation of the HPG axis in others. In patients over 18 years, all but one male had an LH peak >5IU/L, indicating activation of the HPG axis in these adults (Table 2 & 3). We did not note precocious puberty in individuals in this cohort.
![]()
Number of patients (n = 35)
Gender
Mean Age
(years)Age Range
(years)UPD
Total = 16Deletion
Total = 1918
Male
7.5
3.75 - 26
6
12
17
Female
20.5
3.2 - 38
10
7
Table 1: Cohort of 35 individuals with PWS.
![]()
Age (years)
Genetics
Peak LH (IU/L)
Baseline LH
(IU/L)Peak FSH
(IU/L)Baseline FSH
(IU/L)Tanner Breast
14
UPD
0.3
0.3
8.5
5.5
2
14 2/12
UPD
28.3
2.1
15.9
6.1
2
14 4/12
UPD
4.2
0.1
19.5
4.9
3
17
UPD
60.7
3.5
24.5
8
3
21 7/12
UPD
14.7
<2.0
16.3
5.3
3
29 2/12
UPD
15.1
3.2
13.8
9.6
4
29 5/12
UPD
6.95
0.82
12.3
5.21
4
9 5/12
Deletion
2.6
<2.0
9.9
<1.0
1
12 11/12
Deletion
2.8
<2.0
9.8
5.2
1
16 8/12
Deletion
2.3
<2.0
18.5
4.3
2
38
Deletion
26.9
9.1
48.4
34.9
4
Individuals with elevated Peak LH levels indicated in bold
Table 2: LH and FSH values in adolescent and adult females.
![]()
Age (years)
Genetics
Peak LH (IU/L)
Baseline LH
Peak FSH
Baseline FSH
Testicular Size
(IU/L)
(IU/L)
(IU/L)
(cm)
11 1/12
UPD
5.4
<2.0
7.6
1.6
3.5
18 1/6
UPD
9.5
2.1
8.2
5.2
3
18 5/12
UPD
2.8
2.3
4.1
3.8
3
21 1/3
UPD
24.7
20
8.8
3.4
not reported
12 1/6
Deletion
26.3
<2.0
7.5
2.4
2.5
13
Deletion
<2.0
<2.0
3.3
<1.0
1.25
13 3/4
Deletion
37.2
37.2
52.2
52.2
3
15 1/12
Deletion
11.5
3
32.6
23.1
2.5
26 1/4
Deletion
20.7
9.5
73.1
62.7
2
Table 3: LH and FSH values in adolescent and adult males.
In females between 9 and 17 years of age (Table 2), two out of seven had LH levels greater than 5IU/L indicating HPG axis activation and five females had no activation of the HPG axis. Three females in the 9-17 age group with no apparent biochemical activation were noted to have Tanner 2 to 3 breast development, implying either previous estrogen exposure or poor correlation between Tanner staging and HPG axis activation.
An FSH level greater than 30IU/L and an estradiol level less than 60pmol/L is considered indicative of ovarian failure [12].
High LH levels (>30IU/L) are commonly seen in post-menopausal women and may interfere with fertility [13,14].
All of the adult females had peak LH levels >5IU/L demonstrating activation of the HPG axis and expected correlation of Tanner Breast staging that is consistent with puberty. One female had a peak FSH level =48.4IU/L and an estradiol level of 44pmol/L, and another female had a peak LH level of 60.7IU/L and elevated FSH level of 24.5IU/L, suggesting ovarian failure.
In the nine males between 9 and 17 years of age (Table 3), four showed activation of the HPG axis with LH peaks >5IU/L. Two of these males had elevated FSH levels (>18IU/L), with one adult male having an elevated FSH levels of
73.1IU/L consistent with primary gonadal failure [15]. In adult males, three of four had peak LH levels consistent with puberty [15].
Testicular volumes in all males were consistent with the early stages of puberty (2.5cm or greater in the longest dimension). Of note, all adult males had testicular sizes that were smaller than expected for an adult, and one individual had testicular sizes in the pre-pubertal range (<2cm). Testosterone levels were not available for analysis to determine correlation.
We also explored a possible association between the genetic subtypes of the individuals and peak LH levels above and below 5IU/L. There were 22 patients with peak LH <5IU/L, 8 of those patients had UPD and 14 had a deletion.
There were 13 patients with peak LH >5IU/L (indicating activation of the HPG axis), 8 of those patients had UPD and 5 had a deletion (Tables 2 & 3).
Therefore it appears that in the group of patients with peak LH >5IU/L a majority of them were in the UPD group and in the group of patients with peak LH <5IU/L a majority of them were in the deletion group (Figure 1).
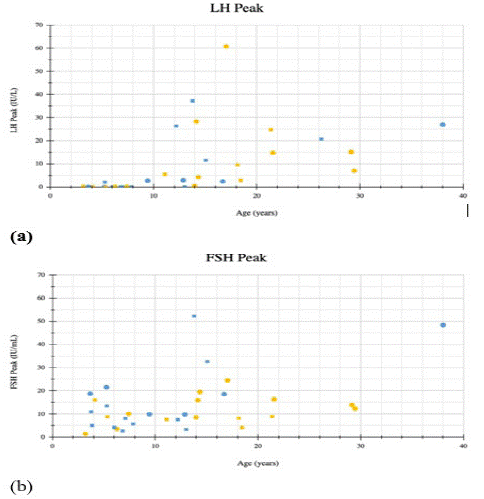
Figure 1: Legend: Square=Males Circle=Females Yellow=UPD Blue=Deletion (a) Peak LH values after gonadorelin administration plotted against age (b) Peak FSH values after gonadorelin administration plotted against age. Dashed line indicates an LH of 5IU/L heralding onset of puberty.
Mean LH levels appear to be higher for PWS females with UPD compared to those with the deletion subtype. Mean LH levels for PWS females with UPD was 13.03+/-19.15IU/L and for those with deletion it was 4.94+/-9.77IU/L, however this was not significant (p-value 0.324) (Table 4).
![]()
Sex
PWS Deletion (over age 9 y., n=8)
PWS UPD (over age 9 y., n=11)
T-Test (Over age 9 y.)
PWS Deletion (all ages, n=19)
PWS UPD (all ages, n=16)
T-Test (all ages)
Sample Size
Mean +/- SD
Sample Size
Mean +/- SD
P Value
Sample Size
Mean +/- SD
Sample Size
Mean +/- SD
P Value
Female
Peak LH
4
8.65 +/-
7
18.61 +/-
0.408
7
4.94 +/-
10
13.03 +/-
0.324
(IU/L)
12.17
20.7
9.77
19.15
Peak
4
21.65
7
15.83 +/-
0.435
7
21.13 +/-
10
12.57 +/-
0.127
FSH
+/-
5.15
14.22
7.06
(IU/L)
18.29
Male
Peak LH
4
23.92
4
10.60 +/-
0.117
12
8.14 +/-
6
7.07 +/-
0.859
(IU/L)
+/-
9.8
12.95
9.36
10.75
Peak FSH (IU/L)
4
41.35
4
7.17 +/-
0.051
12
18.22 +/- 22.68
6
8.90 +/-
0.339
+/- 27.98
2.11
3.89
Table 4: Genetic Analysis of LH and FSH peaks in PWS with UPD vs Deletion.
We ran an analysis of the entire dataset (n=35) and also of the subjects over the age of 9 years (n=19), as expected initiation of puberty is expected to begin at the age of 9 years. Both peak LH and peak FSH levels appear to be higher in males with deletion compared to UPD.
Mean peak FSH level for PWS males in all ages was 18.22+/-22.68 with deletion compared to 8.90+/-3.89 with UPD. Differences were significant only in the peak FSH levels in the >9y. Group p-<0.05) (Table 4), suggesting differences in release of gonadatropins amongst genetic subtypes.
Discussion
Since patients with PWS have other evidence of hypothalamic dysfunction, the dysfunction of their HPG axis has been assumed to be hypothalamic as well. However, prior studies have found mixed results. Hirsch et al. (2015) studied 106 individuals with PWS (49 males and 57 females) [16], and found evidence of hypergonadotropic hypogonadism (primary gonadal failure), with high FSH and low testosterone levels in about half of the adult men. They also found low inhibin B levels (which have a strong positive correlation with sertoli cell function) in most of the men, indicating gonadal dysfunction [17]. Among the group of females, they found normal FSH and LH in over half of the individuals, however, all the adult women had low inhibin B levels, indicating a decrease in ovarian function. Siemensma et. al. studied 61 girls and found that LH and FSH increased after age 10 years as expected and that these girls had an average age of onset of puberty, but had a significantly slower progression to Tanner stage 3 and 4. They also found that maturation of ovarian follicles was impaired, but found no evidence of hypogonadotropic hypogonadism. They hypothesized that girls with PWS may have dysregulation of LH secretion due to the loss of Magel2 resulting in low LH and estradiol levels [18,19]. Mercer and Wevrick studied Magel 2, a candidate gene for PWS, in mice and found that inactivation disrupts circadian rhythm and reduces fertility in both males and females. Irregular estrous cycles in females and decreased testosterone levels in males were found [18].
Recent literature has changed the way we think about reproductive function in those with PWS, but there are still many questions. This is the first study to our knowledge of GnRH stimulation testing for the evaluation of puberty in PWS. While there are reports of precocious puberty in PWS, there were none in this group. Delayed puberty is defined as testicular volume remaining less than 4mL (<2.5cm in diameter) at 14 years in a boy or absence of any breast development at 13 years in a girl [20,21], was seen in half the individuals in our group. In contrast to the findings in Hirsch et. al. [16] where hypergonadotropic hypogonadism was found in about half of the men in the study, our study only had three males who had elevated FSH levels greater than 18IU/L consistent with primary gonadal failure. In females, Tanner breast staging was not sufficient to predict HPG axis activation making laboratory evaluation a necessity in determining true pubertal development. This may be due to difficulty determining staging in individuals with obesity or to initial true breast development with lack of progression, both making physical examination less useful. In males, testicular volumes appear to be smaller than expected for adult males with normal HPG axis function. This may be due to lack of seminiferous tubules during embryonic development or to incomplete pubertal progression. Adults with PWS do appear to have normal response to GnRH stimulation testing after 18 years of age, but it is unclear if this predicts true gonadal function as we do not have testosterone levels or estrogen levels for all patients.
Although we did not find a statistically significant correlation between genetic subtypes (deletion vs UPD) and HPG axis activation, we observed trends within this data. Brandau et. al. (2008) studied gonadal hormone data from 26 females (age 16-50 years) and 24 males (age 16-45 years) with genetically confirmed PWS and found mean LH levels were nearly double for PWS females with UPD compared to PWS females with deletion, though not significantly different [22]. This finding was mirrored in our data set, with mean LH for PWS females with UPD being 13.03+/-19.15IU/L and deletion being 4.94+/-9.77IU/L, however this was not significant (p-value 0.324) (Table 4). We note that we had a small sample size and great degree of variability within our sample. Our study serves as a pilot study and therefore further research analyzing data from a larger sample size is needed to note differences in genetic subtypes. A better understanding of differences between the genetic subtypes holds the potential to allow more personalized medicine with targeted treatments to meet the specific needs of each patient. The more we know about PWS, the better we can educate our patients and their families about the disease, helping them know what to expect when given a diagnosis as well as treatment options.
Lack of phenotype-genotype correlation, inconsistency between physical examination and stimulation testing results and the high variability in age at activation of the HPG axis accentuate the need for the evaluation of pubertal stage in patients with PWS. First morning gonadotropins testing may be used to determine pubertal staging and assess if an individual may benefit from sex-steroid supplementation.
Sex-steroid supplementation may be needed in those who have delayed or incomplete puberty for appropriate growth, bone health and lean body mass. More research into the appropriate timing and impact of sex steroid supplementation is needed. Limited clinical trials exist that have studied sex steroid supplementation in individuals with PWS [23]. It is clear that advanced research is needed to better understand the impact of sex-steroid supplementation on individuals with PWS through the stages of puberty and into their adult lives. Further assessment of pubertal staging in PWS holds the potential to direct the establishment of guidelines for the timing of inducing puberty in individuals with PWS via sex steroid supplementation. In addition to the biological benefits of growth that one gains as they progress through puberty, including increased bone density and lean body mass, there are psychosocial benefits as well.
Without complete physical maturation, an individual may feel socially disconnected amongst their peers of the same age. Therefore, the completion of puberty may lead to more positive mental health outcomes.
Author Statements
Funding
This research was funded by the March of Dimes, grant number U54 HD06122 and National Institute of Health (PI Cassidy).
Informed Consent Statement
Written informed consent was obtained from all subjects involved in the study or their legally responsible caregivers to publish this paper.
Data Availability Statement
All data resulting in this publication is available from vkimonis@uci.edu
Acknowledgments
This research was funded by a grant from the March of Dimes and Birth defects foundation (1999-2003), UC Irvine Institute for Clinical and Translational Science (ICTS). We acknowledge support from the Prader-Willi Syndrome Association (USA) and the Angelman, Rett and Prader-Willi Syndromes Consortium (U54 HD06122) which was part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN) supported through collaboration between the NIH Office of Advancing Translational Science (NCATS) and the National Institute of Child Health and Human Development (NICHD). We thank the GCRC and staff at the University of California, Irvine and Case Western Reserve for their support and help with the patients they looked after. Special thanks go to the families and patients with PWS who freely gave of their time over a prolonged period, many traveling from all over the United States to participate.
Conflicts of Interest
No conflict of interest.
References
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-willi syndrome. Genetics in medicine. 2012; 14: 10-26.
- Bittel DC, Butler MG. Prader–Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert reviews in molecular medicine. 2005; 7: 1-20.
- Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, et al. Prevalence of, and risk factors for, physical ill- health in people with Prader-Willi syndrome: a population-based study. Developmental medicine and child neurology. 2002; 44: 248-55.
- Heksch R, Kamboj M, Anglin K, Obrynba K. Review of Prader-Willi syndrome: the endocrine approach. Translational pediatrics. 2017; 6: 274-285.
- Carrel AL, Myers SE, Whitman BY, Allen DB. Sustained benefits of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome are dose-dependent. Journal of Pediatric Endocrinology and Metabolism. 2001; 14: 1097-106.
- Grugni G, Marzullo P. Diagnosis and treatment of GH deficiency in Prader–Willi syndrome. Best Practice & Research Clinical Endocrinology & Metabolism. 2016; 30: 785-94.
- Burman P, Ritzen EM, Lindgren AC. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocrine reviews. 2001; 22: 787-99.
- Kimonis VE, Tamura R, Gold JA, Patel N, Surampalli A, et al. Early diagnosis in prader–willi syndrome reduces obesity and associated co-morbidities. Genes. 2019; 10: 898.
- Gross-Tsur V, Hirsch HJ, Benarroch F, Eldar-Geva T. The FSH-inhibin axis in prader-willi syndrome: heterogeneity of gonadal dysfunction. Reproductive Biology and Endocrinology. 2012; 10: 39.
- Vogels A, Moerman P, Frijns JP, Bogaert GA. Testicular histology in boys with Prader-Willi syndrome: fertile or infertile?. The Journal of Urology. 2008; 180: 1800-4.
- Kim MS, Hwang PH, Lee DY. A gonadotropin-releasing hormone (GnRH) stimulation test before and after GnRH analogue treatment for central precocious puberty: has the gnrh test been adequately simplified?. The Indian Journal of Pediatrics. 2015; 82: 996-1000.
- Baird DT. Amenorrhoea. The Lancet. 1997; 350: 275-9.
- Wu AH. Tietz clinical guide to laboratory tests-E-book. Elsevier Health Sciences. 2006.
- ESHRE TT. ASRM-Sponsored PCOS Consensus Workshop Group. Consensus on infertility treatment related to polycystic ovary syndrome. Fertility and sterility. 2008; 89: 505-22.
- Gordetsky J, van Wijngaarden E, O’Brien J. Redefining abnormal follicle-stimulating hormone in the male infertility population. BJU international. 2012; 110: 568-72.
- Hirsch HJ, Eldar-Geva T, Bennaroch F, Pollak Y, Gross-Tsur V. Sexual dichotomy of gonadal function in Prader–Willi syndrome from early infancy through the fourth decade. Human Reproduction. 2015; 30: 2587-96.
- Meachem SJ, Nieschlag E, Simoni M. Inhibin B in male reproduction: pathophysiology and clinical relevance. European journal of endocrinology. 2001; 145: 561-71.
- Siemensma EP, van Alfen-van der Velden AA, Otten BJ, Laven JS, Hokken-Koelega AC. Ovarian function and reproductive hormone levels in girls with Prader-Willi syndrome: a longitudinal study. The Journal of Clinical Endocrinology & Metabolism. 2012; 97: E1766-73.
- Mercer RE, Wevrick R. Loss of magel2, a candidate gene for features of Prader-Willi syndrome, impairs reproductive function in mice. PLoS One. 2009; 4: e4291.
- Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Archives of disease in childhood. 1969; 44: 291-303.
- Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Archives of disease in childhood. 1970; 45: 13-23.
- Brandau DT, Theodoro M, Garg U, Butler MG. Follicle stimulating and leutinizing hormones, estradiol and testosterone in Prader–Willi syndrome. American journal of medical genetics. Part A. 2008; 146: 665.
- Noordam C, Höybye C, Eiholzer U. Prader–willi syndrome and hypogonadism: A review article. International Journal of Molecular Sciences. 2021; 22: 2705.