
Case Report
J Pediatri Endocrinol. 2023; 8(1): 1060.
Endocrine Complications in an Adolescent with Fibrous Dysplasia
Look SE*; Timme K
Department of Pediatrics, University of Utah, USA
*Corresponding author: Look, SE Department of Pediatrics, University of Utah, 81 N Mario Capecchi Dr, Salt Lake City, UT 84113, USA. Tel: 239-699-2406 Email: sydney.look@hsc.utah.edu
Received: September 28, 2023 Accepted: October 20, 2023 Published: October 27, 2023
Abstract
McCune Albright Syndrome is characterized by the triad of fibrous dysplasia, cutaneous manifestations, and endocrine dysfunction. While this disease is typically associated with precocious puberty, there are other endocrinopathies to consider. An adolescent with fibrous dysplasia was diagnosed with acromegaly, hyperthyroidism, phosphate wasting, and hypogonadotropic hypogonadism. His course was complicated by optic neuropathy and parasellar mass ultimately determined to be due to progression of skull base fibrous dysplasia. There was additional consideration of side effects from octreotide. This case exemplifies the potential for under-recognition of acromegaly in McCune Albright Syndrome, which can be associated with cranial nerve deficits. Fibrous dysplasia progression may be associated with additional endocrine dysfunction depending on the location, with potential for necrosis or structural damage of the pituitary gland. Physicians should have high suspicion for endocrinopathies with low threshold for evaluation to optimize outcomes in this population.
Keywords: Acromegaly; McCune albright syndrome; Optic neuropathy; Octreotide
Abbreviations: MAS: McCune Albright Syndrome; FD: Fibrous Dysplasia; GNAS1: Guanine Nucleotide Binding Protein, Alpha Stimulating Activity Polypeptide-1; GH: Growth Hormone; IGF-1: Insulin-Like Growth Factor-1; NR: Normal Range; TSH: Thyroid Stimulating Hormone; T4: Tyroxine; T3: Triiodothyronine; LH: Luteinizing Hormone; FSH: Follicle-Stimulating Hormone; MRI: Magnetic Resonance Imaging
Introduction
MAS is characterized by FD, cutaneous manifestations including café-au-lait macules, and endocrine dysfunction. MAS occurs due to a mosaic GNAS1 gene mutation with a variety of clinical effects [1-4]. There are multiple endocrinopathies in MAS, though GNAS1 mutations are typically associated with endocrine hyperfunction with precocious puberty as one of the more common manifestations. Acromegaly is also common in MAS (20-30%) with an association in patients with skull base lesions [3,5,6].
Acromegaly is caused by excess GH production from the pituitary gland, which stimulates IGF-1 and promotes rapid dysregulated physical growth. GH excess in childhood, known as gigantism, results in late closure of epiphyses, often manifesting as tall stature. Acromegaly occurs after growth plates fuse and results in coarse facial features and gradual enlargement of hands and feet along with systemic effects. While acromegaly is often caused by a GH-secreting pituitary adenoma, it can also occur due to diffuse somatotroph hyperplasia as part of a genetic syndrome [6,7]. Medical treatment occurs if transsphenoidal surgery is ineffective or contraindicated. Medications include somatostatin analogues that inhibit GH secretion such as octreotide (injection or oral formulation), GH receptor antagonists, and rarely dopamine antagonists.
Case Presentation
A 17-year-old male with FD was referred to pediatric endocrinology for evaluation of abnormal thyroid function noted during an emergency department visit for fatigue and palpitations. His exam was remarkable for tall stature, macrocephaly, coarse facial features, and large hands and feet.
He fractured his right femur at age 5 leading to diagnosis of FD. He has a limb length discrepancy and uses crutches and a wheelchair for mobility. He has scoliosis and an acquired Chiari 1 malformation. He reported normal pubertal timing with exam consistent with pubertal stage 5. He had hyperpigmentation on his back determined to be reaction to surgical tape but no distinct café au lait macules. His height was 6 feet 1.8 inches (187.4cm), while his sex-adjusted mid-parental height was 5 feet 10 inches (178cm) (Figure 1). In comparison, his actual height was greater than 2 standard deviations above his anticipated adult height.
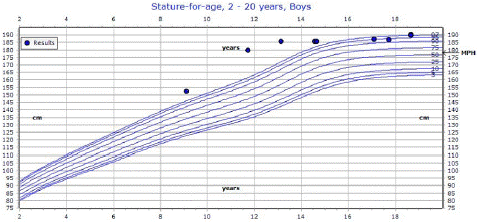
Figure 1: Patient’s growth chart with mid-parental height. At initial endocrine evaluation, patient was 187.4 cm, at the 95th percentile for age and sex. Final adult height is shown at 190 cm, at the 97th percentile for age and sex. Mid-parental height is calculated at 178 cm. Of note, patient’s height was > 99th percentile in early adolescence.
Laboratory evaluation confirmed clinical suspicion for GH excess. His IGF-1 was elevated at 786ng/mL (+3 Standard Deviations) with failure to suppress GH on oral glucose tolerance test. He was also evaluated for other endocrinopathies. He had a low TSH of 0.01mcIU/mL (NR 0.5–5.0mcIU/mL), with normal free T4 of 1.26ng/dL (NR 0.73-1.57ng/dL) and total T3 of 138ng/dL (NR 90-168ng/dL). He had low testosterone at 24ng/dL (NR 148-794ng/dL) with low LH of 0.2 munit/mL (NR 1.1-9.0munit/mL) and low normal FSH of 1.1mIU/mL (NR 0.9-7.8mIU/mL). His prolactin level was mildly elevated at 19.2ng/mL (NR 2.0-14.0ng/mL). He had low serum phosphorous at 2.6mg/dL (NR 2.9-5.0mg/dL). His fractionated excretion of phosphate was elevated at 24% (NR 10-20%). A baseline MRI brain did not show a pituitary adenoma but did confirm extensive skull FD.
The clinical diagnosis of MAS was made based on FD, hyperpigmentation, and endocrine dysfunction with GH excess, subclinical hyperthyroidism, hypogonadotropic hypogonadism, and renal phosphate wasting. He started octreotide injections for acromegaly. He started phosphate supplements and calcitriol for hypophosphatemia and phosphate wasting. He did not require treatment for subclinical hyperthyroidism. Testosterone supplementation for hypogonadotropic hypogonadism was deferred until growth factors were optimized.
Six months after starting octreotide, the patient developed acute headache and dizziness prompting evaluation in the emergency department. He had an unreactive left pupil, left 3rd and 6th nerve palsies, and decreased left-sided vision. An MRI showed interval development of a sellar mass. There were higher attenuation areas suspicious for intrinsic bleeding.Surrounding bony destructive changes were worsened in comparison to previous imaging. The imaging correlated with his symptoms, however the etiology remained unclear. Repeat MRI with angiography (Figure 2) favored parasellar focal degeneration and necrosis of FD with secondary hemorrhage causing mass effect, although pituitary adenoma with hemorrhage could not be excluded.
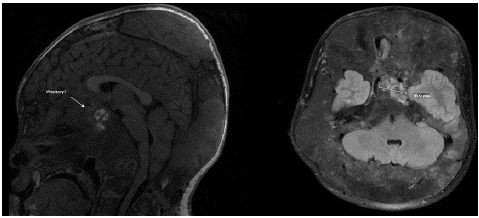
Figure 2: Patient’s MRI demonstrates likely parasellar/basisphenoid focal degeneration and necrosis of fibrous dysplasia with areas of intrinsic enhancement suggestive of secondary hemorrhage into the lesion. There is extensive skull base fibrous dysplasia with crowding of intracranial contents.
The patient was hospitalized and initiated on dexamethasone. Due to reports of spontaneous remission of GH excess due to pituitary apoplexy in patients on octreotide [8,9], octreotide was held while growth factors were monitored. Repeat imaging and serial lab monitoring demonstrated a normal pituitary gland and stable pituitary function, lowering suspicion for apoplexy. He was discharged from the hospital following improvement of headache and dizziness with a dexamethasone taper and close neurosurgical and ophthalmologic follow-up.
When his growth factors rose outpatient, octreotide injections were restarted. One month later, the patient developed acute peripheral edema. His work-up was reassuring against heart failure with normal echocardiogram, nephrotic syndrome with normal urinalysis, and thrombosis with normal ultrasounds and angiogram. Additional considerations included mixed dermatitis, myxoma associated with FD, or cellulitis. It was unclear if this was related to a recent increased dose of octreotide injections from 20 to 30 mg daily. Edema due to octreotide injection is listed as a rare adverse effect and considered a diagnosis of exclusion. The patient was hesitant to continue injections and transitioned to daily oral octreotide. He was treated with supportive wound care and his edema resolved.
Over the following year, the patient had persistent severe left-sided vision loss. He developed progressive loss of vision in his right eye prompting right optic nerve decompression. His endocrine care was transitioned to adult endocrinology. He developed clinical hyperthyroidism and started methimazole. He also started testosterone supplementation for hypogonadotropic hypogonadism.
Discussion
This case is impactful because the patient’s GH excess was likely undiagnosed for years. GH excess may be masked in MAS secondary to craniofacial FD obscuring what would otherwise be pathognomonic coarse facial features. This case illustrates the importance of evaluating stature in the context of familial patterns, as the patient was significantly taller than his anticipated adult height.
Physicians caring for patients with skull base FD should be cognizant of evaluating for GH excess with a low threshold for work-up due to potential sequelae. This patient developed vision loss due to FD progression. Vision and hearing deficits have been identified as more common in patients with FD and GH excess (4% vs 33%) [5]. It has been suggested that GH excess exacerbates FD, especially at the craniofacial level, and earlier diagnosis and treatment of GH excess may prevent co-morbidities such as optic neuropathy [10]. Early recognition of vision or hearing deficits is important for prompt neurosurgical and ophthalmologic treatment to optimize function.
This case is unique because his acute presentation with optic neuropathy was associated with initial concern for pituitary apoplexy related to octreotide therapy, though it was determined that his symptoms were likely related to FD progression and necrosis. This presents a challenge for physicians to navigate, as FD progression can lead to side effects that complicate treatment. This is seen in this case with diagnosis of hypogonadotropic hypogonadism. This was identified after normal puberty development and timing, suggesting acquired dysfunction during later adolescence. We hypothesize FD progression with associated necrosis led to gonadotroph cell damage over time. There are similar cases in literature [11,12], which suggest physicians should also evaluate for endocrine hypofunction.
References
- Boyce AM, Collins MT. Fibrous dysplasia/McCune-Albright syndrome: A rare, mosaic disease of Ga s activation. Endocr Rev. 2020; 41: 345-70.
- Boyce AM, Florenzano P, de Castro LF, et al. Fibrous dysplasia / McCune-Albright syndrome. 2015 Feb 26. In: Seattle: University of Washington. Seattle. 1993-2023; Updated 2019 Jun 27. GeneReviews® [internet] Adam MP, Mirzaa GM, Pagon RA, et al., editors. Available from: https://www.ncbi.nlm.nih.gov/books/NBK274564/.
- Javaid MK, Boyce A, Appelman-Dijkstra N, Ong J, Defabianis P, Offiah A, et al. Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: a consensus statement from the FD/MAS international consortium. Orphanet J Rare Dis. 2019; 14: 139.
- Spencer T, Pan KS, Collins MT, Boyce AM. The clinical spectrum of McCune-Albright syndrome and its management. Horm Res Paediatr. 2019; 92: 347-56.
- Akintoye SO, Chebli C, Booher S, Feuillan P, Kushner H, Leroith D, et al. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J Clin Endocrinol Metab. 2002; 87: 5104-12.
- Bhansali A, Sharma BS, Sreenivasulu P, Singh P, Vashisth RK, Dash RJ. Acromegaly with fibrous dysplasia: McCune-Albright syndrome -- clinical studies in 3 cases and brief review of literature--. Endocr J. 2003; 50: 793-9.
- Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab. 2014; 99: 1955-69.
- Fraser LA, Lee D, Cooper P, Van Uum S. Remission of acromegaly after pituitary apoplexy: case report and review of literature. Endocr Pract. 2009; 15: 725-31.
- Kumar S, Sharma S. Pituitary apoplexy causing spontaneous remission of acromegaly following long-acting octreotide therapy: rare drug side effect or just a coincidence. Oxf Med Case Rep. 2016; 2016: 81-3.
- Boyce AM, Glover M, Kelly MH, Brillante BA, Butman JA, Fitzgibbon EJ, et al. Optic neuropathy in McCune-Albright syndrome: effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab. 2013; 98: E126-34.
- Lima-Martínez MM, Gil V, Mederico M, Gómez-Pérez R. Hypogonadotropic hypogonadism in a man with McCune Albright syndrome. Endocrinol Nutr. 2013; 60: 145-7.
- Shires R, Whyte MP, Avioli LV. Idiopathic hypothalamic hypogonadotropic hypogonadism with polyostotic fibrous dysplasia. Arch Intern Med. 1979; 139: 1187-9.