
Case Report
J Pediatri Endocrinol. 2023; 8(2): 1062.
Focal Congenital Hyperinsulinism (CHI) Due To Paternally Inherited Variant in ABCC8 Gene: Importance of Genetic Testing & PET Imaging in Stratification & Management of Diazoxide Unresponsive CHI
Abhishek Kulkarni¹*; Joewin Monteiro²; Devika Desai³
1Senior Consultant Paediatric and Adolescent Endocrinologist, SRCC Children’s Hospital, Mumbai & Sir HN Reliance Foundation Hospital, Mumbai, India
2Clinical and Research Fellow, Department of Pediatric and Adolescent Endocrinology, SRCC Children’s Hospital, Haji Ali, Mumbai, India
3Clinical Associate, Department of Paediatric Endocrinology, SRCC Children’s Hospital, Mumbai, India
*Corresponding author: Abhishek Kulkarni Senior Consultant Paediatric and Adolescent Endocrinologist, SRCC Children’s Hospital, Mumbai & Sir HN Reliance Foundation Hospital, Mumbai, India. Email: cpedndc@gmail.com
Received: October 30, 2023 Accepted: November 25, 2023 Published: December02, 2023
Abstract
Congenital Hyperinsulinism (CHI) may be transient or permanent and inherited either in autosomal recessive or dominant forms. Variants in ABCC8 and KCNJ11 genes coding for Sulphonyl Urea Receptor (SUR1) and potassium channel subunit (Kir6.2) respectively, account for 82% cases of diazoxide-unresponsive patients and 45% of all cases of CHI. The histology of CHI is classified into focal and diffuse. Approximately half of all cases can be classified as diffuse, 40 % as focal, and 10% of cases are considered atypical. We report a 1 month old male with recurrent hypoglycemia detected with focal congenital hyperinsulinism secondary to a paternally inherited heterozygous ABCC8 gene mutation. Focal disease results due to inheritance of a monoallelic recessive paternal ABCC8 or KCNJ11 variant together with somatic Loss of Heterozygosity (LOH) of the maternal allele at 11p15 within a pancreatic progenitor cell. Infants & children with focal disease may be cured by resection of their hyperplastic lesion and the risk for postoperative diabetes is significantly lower as compared to that following pancreatectomy for diffuse CHI. The report describes molecular mechanisms underlying CHI & emphasises the need of genetic testing and diagnostic functional imaging modalities in diazoxide unresponsive CHI to guide management and improve clinical outcomes.
Keywords: Focal congenital hyperinsulinism; CHI; Recessively inherited monoallelic mutation; 18F-DOPA PET
Introduction
Congenital Hyperinsulinism (CHI) is a rare, clinically heterogenous disorder and the most frequent cause of persistent hypoglycaemia in neonates, infants, and children. The incidence of CHI varies between 1 in 50,000 live births to as high as 1 in 2500 live births in countries with high degree of consanguinity. The etiology of CHI is largely genetic. Mutations in at least 12 genes that play a role in regulating beta-cell insulin secretion are implicated in the pathogenesis of HI. Approximately 50% of diazoxide-responsive cases and 10% of diazoxide-unresponsive cases of CHI have unknown etiology, suggesting that additional genes & genetic mechanisms may need elucidation [1].
The histology of CHI is classified into focal and diffuse. Approximately half of all cases can be classified as diffuse, 40 % as focal, and 10% of cases are considered atypical [2]. Genetic mutation testing is well established as standard of care to define best approaches to treatment of CHI especially in diazoxide unresponsive patients. Fluorine-18L-3,4-hydroxyphenylalanine positron emission tomography (18F-DOPA-PET) is considered the gold standard for preoperative differentiation of focal from diffuse disease and localization of the focal lesion [2]. We report a 1 month old male with recurrent hypoglycemia detected with focal congenital hyperinsulinism secondary to a paternally inherited heterozygous ABCC8 gene mutation.
Case Report
A 1 month old male, born at term by LSCS of a non-consanguineous union was referred with history of multiple episodes of hypoglycaemic seizures since day 4 of life. A critical sample was sent on presentation at capillary blood glucose value of 40 mg/dl which depicted an insulin level of 5.7 mIU/ml for a corresponding laboratory RBS value of 35 mg/dl. The values of cortisol, GH, ammonia, lactate as sent from the critical sample were within permissible limits & beta hydroxybutyrate was undetectable. Screening for sepsis & IEMs were negative. Inability to wean the baby off a Glucose Infusion Rate of >6mg/kg/min inspite of use of threshold doses of diazoxide & hydrochlorothiazide suggested unresponsiveness to diazoxide. The baby was initiated on subcutaneous octreotide therapy & was weaned off IV fluids over the next 48 hours. Euglycemia was maintained with a subcutaneous dose of 5 mcg/kg octreotide administered 8 hourly & timely breast feeds.
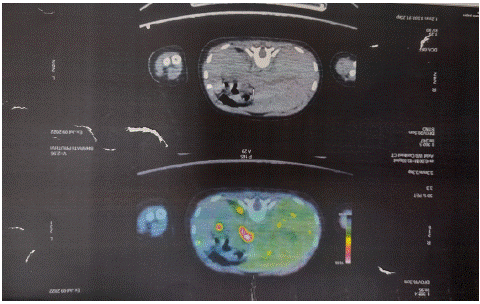
Figure 1:
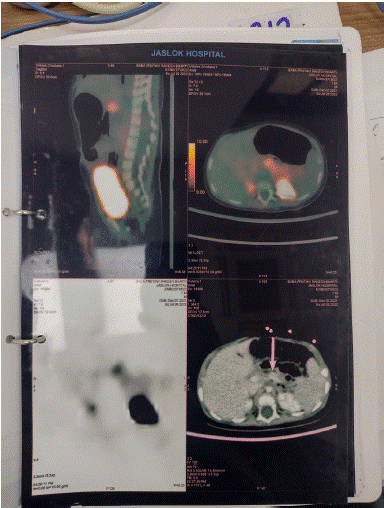
Figure 2:
Targeted exome analysis yielded a heterozygous deletion in c1715del (p.ser572CysfsTer33) in exon 12 of the ABCC8 gene which was confirmed by sanger sequencing. Parental segregation analysis confirmed presence of the variant in the father. 18F-DOPA PET depicted focal abnormal dopaminergic concentration in proximal part of the body of pancreas with SUV max 11.2 vs SUV max of rest of pancreas 8.33 which was reaffirmed by focal increased GLP1 receptor expression at the site by 68 Ga EXENDIN PET CT.
Enucleation and excision of the lesion was performed after confirmation of the site by intra operative ultrasound. The lesion was palpable at the junction of the body and head of the pancreas. Histopathology depicted well differentiated insulin expressing cells involving the entire bit submitted encapsulated in basement membrane of collagen fibre bundles. Excision led to cessation of requirement of octreotide once the infant was weaned off IV fluids to exclusive breastfeeding. Since then the infant has remained euglycemic consistent with plausible remission or cure subsequent to the lesionectomy. Neurodevelopmental surveillance for sequelae of prior episodes of hypoglycemia and recurrence of lesion(s) is being maintained.
Discussion
Congenital Hyperinsulinism (CHI) may be transient or permanent and inherited either in autosomal recessive or dominant forms. Variants in ABCC8 and KCNJ11 genes coding for Sulphonyl Urea Receptor (SUR1) and potassium channel subunit (Kir6.2) respectively, account for 82% cases of diazoxide-unresponsive patients and 45% of all cases of CHI [3]. It is stratified into diffuse and focal CHI histologically & both of these may be unresponsive to diazoxide treatment and difficult to delineate clinically. Diffuse disease is usually inherited recessively with abnormal pancreatic β-cells throughout the pancreas. Atypical diffuse forms exist due to mosaic Uniparental Disomy (UPD) in patients with dominantly inherited ABCC8 or KCNJ11 gene variants [4]. In focal forms, the disease is confined to an isolated lesion in the pancreas and results due to inheritance of a monoallelic recessive paternal ABCC8 or KCNJ11 variant together with somatic Loss of Heterozygosity (LOH) of the maternal allele at 11p15 within a pancreatic progenitor cell. Infants & children with focal disease may attain cured by resection of their hyperplastic lesion and the risk for postoperative diabetes is significantly lower as compared to that following pancreatectomy for diffuse CHI.
The presence of a monoallelic recessive K+ ATP variant is reported to predict focal CHI with high sensitivity (97%) and specificity (90%) [5]. Diagnosis of focal CHI must be based on clinical features combined with the results of genetic testing and 18F-DOPA PET-CT scanning. Microsatellite analysis of the resected pancreatic tissue depicts maternal LOH in the region spanning chromosome 11p15.5-11p15.1, which includes the ABCC8 locus, unmasking a recessive disorder [3]. LOH most likely occurs either in the late embryonic or early neonatal period & the size of the focal lesion depends on the its timing. Prior knowledge of the genetic etiology can guide evaluations, surgical procedures, inform prognosis and aid genetic counselling. The availability of rapid next generation gene sequencing and 18F-DOPA PET-CT imaging add immense value to management of diazoxide-unresponsive CHI. Although treatment for CHI has improved over the last decade, the risk of adverse neurodevelopment remains high with incidence frequencies varying between 26% and 48% in contemporary literature [3].
Conclusion
Our case report describes the molecular mechanisms underlying CHI & emphasises the need of genetic testing and diagnostic functional imaging modalities in diazoxide unresponsive CHI to guide management and improve clinical outcomes.
References
- Lord K, De León DD. Monogenic hyperinsulinemic hypoglycemia: current insights into the pathogenesis and management. Int J Pediatr Endocrinol. 2013; 2013: 3.
- Gopal-Kothandapani JS, Hussain K. Congenital hyperinsulinism: role of fluorine-18L-3, 4 hydroxyphenylalanine positron emission tomography scanning. World J Radiol. 2014; 6: 252-60.
- Joyce CM, Houghton JA, O’Halloran DJ, O’Shea PM, O’Connell SM. Inheritance of a paternal ABCC8 variant and maternal loss of heterozygosity at 11p15 retrospectively unmasks the etiology in a case of Congenital hyperinsulinism. Clin Case Rep. 2020; 8: 1217-22.
- Hussain K, Flanagan SE, Smith VV, Ashworth M, Day M, Pierro A, et al. An ABCC8 gene mutation and mosaic uniparental isodisomy resulting in atypical diffuse congenital hyperinsulinism. Diabetes. 2008; 57: 259-63.
- Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013; 98: E355-63.