
Case Report
J Pediatr & Child Health Care. 2017; 2(1): 1014.
Progressive Familial Intrahepatic Cholestasis Type 3: A Case Report and Literature Review
Azami MA¹*, Lahbali O¹, Lamalmi N¹, Oukabli M² and Bouzidi AA²
¹Department of pathology, Child Hospital in Rabat, Mohammed V University, Morocco
²Department of pathology, Mohamed V Military Hospital, Mohammed V University, Morocco
*Corresponding author: Mohamed Amine Azami, Department of Pathology, Child Hospital in Rabat, Mohammed V University in Rabat, Morocco
Received: May 05, 2017; Accepted: June 02, 2017; Published: June 09, 2017
Abstract
Progressive Familial Intrahepatic Cholestasis (PFIC) is a group of rare disorders which are caused by defect in bile secretion and present with intrahepatic cholestasis, usually in infancy and childhood.
Based on clinical presentation, laboratory findings, liver histology and genetic defect, these are broadly divided into three types: PFIC type 1(Byler’s disease), PFIC type 2 and PFIC type 3.
The main clinical presentation is in the form of cholestatic jaundice and pruritus. Serum Gamma Glutamyl Transpeptidase (GGT) is normal in patients with PFIC1/2 while it is raised in patients with PFIC3.
PFIC can progress rapidly and cause cirrhosis during infancy or may progress relatively slowly with minimal scarring well into adolescence. Few patients have survived into the third decade of life without treatment.
In PFIC3, clinical signs of cholestasis are noted within the first year of life in about one third of patients and rarely in the neonatal period, in contrast to PFIC1 and 2.
We report in this paper a case of a 45-days old boy presented early in the infantile period with deep jaundice, his investigations showed progressive cholestatic jaundice, high liver enzymes and high GGT. Hepatitis and metabolic errors were excluded. The liver biopsy showed a prominent parenchymal bile stasis without features of bile obstruction or an evidence of paucity of bile ducts. These findings are going with the diagnosis Progressive Familial Intrahepatic Cholestasis (PFIC3).
Keywords: PFIC3; Liver disease; Cholestasis; MDR3 deficiency
Introduction
Progressive Familial Intrahepatic Cholestasis type 3 (PFIC3) is an autosomal recessive disorder of cholestasis of hepatocellular origin. The onset of PFIC3 is typically in infancy or in childhood.
PFIC3 has been ascribed to a defect in MDR3 (class III multidrug resistance p-glycoprotein) or ABCB4 gene, and the current preferred terminology is MDR3 deficiency [1].
Children with PFIC3 often present in the first year of life with clinical signs of cholestasis that leads to progressive liver disease and cirrhosis. PFIC3 patients have elevated serum Gamma Glutamyl Transferase activity (GGT) that differentiates them from those with PFIC 1 and 2.
Liver histology shows hepatocellular damage, inflammatory infiltration of the portal tract and fibrosis. In advanced disease, extensive fibrosis and biliary cirrhosis are seen [2].
We recently encountered a new presentation of this rare entity and share our experience in its diagnosis and treatment with progression of disease.
Case Report
45-days-old boy is a product of a full term normal delivery with birth weight of 2800 grams, pregnancy was uneventful, and his post-natal period passed without problems. His symptoms started at the first month of life when the parent noticed that he has yellow discoloration of skin and sclera which was becoming deeper by time then he started to have severe itching to his skin. His parents are first degree cousins but no family history of similar condition. On physical examination the child was deeply jaundiced with multiple scratch marks all over his body. His weight was 3.8 kg, his height was 59cm with moderate hepatosplenomegaly. There was no ascites, and no other signs of chronic liver disease or rickets. The results of his investigations showed a normal CBC, urea, creatinine and electrolytes. Total bilirubin was 19.2 gm with a direct of 7.7 gm. His liver enzymes, alkaline phosphatase 552, AST: 509, ALT: 231, Gama Glutamyl Transpeptidase (GGT):541 IU/l (normal:7-32). Prothrombin time was 14 seconds (control=14seconds) and partial thromboplastin time was 30seconds (control 32 seconds), albumin, cholesterol and triglycerides levels were normal. Infectious screen for hepatitis A, B, C, and TORCH infections were all negative. TSH and T4 are normal. Abdominal ultrasound showed slightly increased echogenicity of the enlarged liver. Liver biopsy showed prominent parenchymal bile stasis and partially distorted architecture but no features of bile obstruction or paucity of bile ducts (Figure1 and Figure 2). The patient is on medical treatment including fat soluble vitamins (A,D,E,K) and phenobarbitone without proper response and he is still having severe pruritis and deterioration of liver function so he will be referred for surgical treatment.
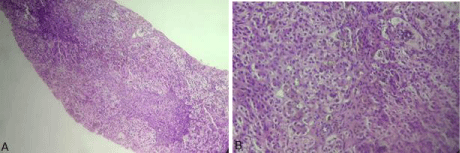
Figure 1: Liver biopsy showing bile duct plugs along with multinuclear giant/
hydropic cells with moderate portal inflammation and fibrosis.( Hematoxylin
and eosin stain ; A: 100x, B: 200x).
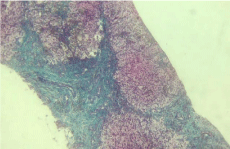
Figure 2: Liver biopsy showing micro-nodular cirrhosis with fibrous portal
expansion and inter-portal bridging. (Masson trichrome X100).
Discussion
Progressive Familial Intrahepatic Cholestasis (PFIC) is a heterogeneous group of liver disorders of autosomal recessive inheritance, presenting as intrahepatic cholestasis in infancy or early childhood and resulting in End Stage Liver Disease (ESLD) and death or liver transplantation in infancy to adulthood [3,4].
Based on clinical presentation, laboratory findings, liver histology and genetic defect, these are broadly divided into three types: PFIC type 1(Byler’s disease), PFIC type 2 and PFIC type 3 [5].
All the three types of PFIC are caused by defects in bile secretion from hepatocyte to canaliculi. The defects are in form of penetrant mutations in genes encoding proteins associated with hepatocellular transport system [6].
The PFIC3 is different from PFIC1 and 2 in clinical presentation and is associated with high Gamma Glutamyl Transpeptidase (GGT) as compared to normal/low GGT in patients with type 1 and 2 [7,8]. It is caused by defects in Adenosine triphosphate-binding cassette, subfamily B, member 4 (ABCB4) gene encoding Multi Drug Resistance class III (MDR3) protein, located on chromosome7 (7q21) [3].
In PFIC3, clinical signs of cholestasis are noted within the first year of life in about one third of patients and rarely in the neonatal period, in contrast to PFIC1 and2. PFIC3 may also manifest later in infancy, in childhood or even in young adulthood. Pruritusis usually mild. Evolution is characterized by chronicicteric or anicteric cholestasis, portal hypertension and liver failure. In half of the patient, liver transplantation is required at a mean age of 7.5 years. No liver tumor has yet been reported in association with PFIC3 [4].
At Laboratory findings patients with PFIC1 and PFIC2 have normal serum Gamma-Glutamyl Transferase (GGT) activity, normal serum cholesterol level and very high serum bile acid concentration. However Patients with PFIC3 have a persistent high serum GGT activity (that differentiates them from those with PFIC 1 and 2), normal serum cholesterol level and moderately raised concentrations of serum primarybile salts [9-12].
Ultrasonography is the first test which is essentially normal except for the presence of cholelithiasis in some cases of PFIC 3. It also helps to exclude other causes of extra hepatic cholestasis. Cholangiography helps in excluding sclerosing cholangitis in patients with high GGT cholestasis [13].
Biopsy pathology varies with age at diagnosis. In PFIC3, liver histology, obtained at the time of diagnosis, shows portal fibrosis and true ductular proliferation with mixed inflammatory infiltrate. In a few instances, cholestasis is present in the lobule and in some ductules containing bile plugs. Slight giant transformation of hepatocytes can be observed. Cytokeratin immunostaining confirms the strong ductular proliferation within the portal tract. At a later stage there is extensive portal fibrosis and a typical picture of biliary cirrhosis. Interlobular bile ducts are seen in most portal tracts and there is neither periductal fibrosis nor biliary epithelium injury [14].
Immunostaining of the MDR3 protein can be variable, ranging from complete absence to diminished staining, depending on the mutation. The diagnosis of PFIC3 is confirmed by molecular genetic analysis of the ABCB4 gene [15].
Medical therapy is the first line of treatment in patients with all types of PFIC. The objectives are to provide relief from pruritus, improve the nutritional status, correct vitamin deficiencies and treat complications of advanced liver disease like ascites and variceal bleeding if present [16].
The most commonly used drug for pruritus is Urso-Deoxy Cholic Acid (UDCA) which is a hydrophilic bile acid, non-toxic to hepatocytes [17].
If these therapies fail, liver transplantation represents the only alternative and should be considered in patients with ESLD (End Stage Liver Disease), HCC (Hepato Cellular Carcinoma) or those with poor quality of life due to refractory pruritus despite medical treatment and biliary diversion. Liver transplantation improves cholestasis and its symptoms in 75-100% patients, irrespective of PFIC subtype over a short term follow-up of 3-5 years [18].
Conclusion
Progressive Familial Intrahepatic Cholestasis type 3 (PFIC3) is an autosomal recessive disorder of cholestasis of hepatocellular origin. It is caused by defects in Adenosine triphosphate-binding cassette, subfamily B, member 4 (ABCB4) gene encoding Multi Drug Resistance class III (MDR3) protein.
Few patients have survived into the third decade of life without treatment. Liver transplantation represents the only alternative, but unfortunately is not always available. In the future, therapies such as cell, gene or specific targeted pharmacological therapies, might represent an alternative therapy for all types of PFIC.
References
- Deleuze JF, Jacquemin E, Dubuisson C, Cresteil D, Dumont M, Erlinger S, et al. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology. 1996; 23: 904-908.
- Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2008; 46: 241-252.
- Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009; 4: 1.
- Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012.
- Russell DW. The enzymes, regulation and genetics of bile acid synthesis. Annu Rev Biochem 2003; 72: 137-74.
- Bull LN, Carlton VE, Stricker NL, Baharloo S, DeYoung JA, Freimer NB, et al. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC 1] and Byler syndrome): evidence for heterogenicity. Hepatology. 1997; 26: 155-164.
- Jansen PL, Muller M. Genetic cholestasis: lessons from the molecular physiology of bile formation. Can J Gastroenterol. 2000; 14: 233-238.
- De vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998; 95: 282-287.
- van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin Liver Dis. 2001; 21: 535-544.
- Thompson R, Strautnieks S. BSEP: function and role in progressive familial intrahepatic cholestasis. Semin Liver Dis. 2001; 21: 545-550.
- Chen HL, Chang PS, Hsu HC, Ni YH, Hsu HY, Lee JH, et al. FIC1 and BSEP defects in Taiwanese patients with chronic intrahepatic cholestasis with low gama-glutamyltranspeptidase levels. J Pediatr. 2002; 140: 119-124.
- Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol. 2003; 39: 447-452.
- Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014; 4: 25-36.
- Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009; 4: 1.
- Ramraj R, Finegold MJ, Karpen SJ. Progressive familial intrahepatic cholestasis type 3: overlapping presentation with Wilson disease. Clin Pediatr. 2012; 51: 689-691.
- Feranchak AP, Ramirez RO, Sokal RJ. Medical and NutritionalManagement of Cholestasis. Williams and Wilkins. 2001.
- Poupon R. Intrahepatic cholestasis of pregnancy: from bedside to bench to bedside. Liver Int. 2005;25:467-468.
- Soubrane O, Gauthier F, DeVictor D, Bernard O, Valayer J, Houssin D, et al. Orthotopic liver transplantation for Byler disease. Transplantation. 1990; 50: 804-806.