
Review Article
J Pediatr & Child Health Care. 2022; 7(1): 1054.
Charcot-Marie-Tooth Disease 4G Caused by Homozygous Deletion of Exon 4 in HK1 Gene due to Inbreeding: A Case Report and Literature Review
Xu H#, Ying G#, Qian H#, Wang S, Huang T, Chen J, Zhu X, Guo H, Zheng G and Zhang G*
Department of Neurology, Children’s Hospital of Nanjing Medical University, China
#These authors contributed equally to this article
*Corresponding author: Gang Zhang, Department of Neurology, Children’s Hospital of Nanjing Medical University, No.72 Guangzhou Road, Nanjing, Jiangsu, China
Received: July 13, 2022; Accepted: August 08, 2022; Published: August 15, 2022
Abstract
Objectives: The objective is to investigate the clinical and genotypic characteristics of Charcot-Marie-Tooth disease, caused by HK1 gene mutation.
Methods: The detailed medical history of the child was collected and the clinical symptoms were summarized. The genomic DNA was extracted from the 2ml of peripheral blood of child and their parents, and the whole exome sequencing was performed, and the related literatures were reviewed.
Results: A 7-year-old girl with unstable walking for 7 months, left claudication, obvious valgus of left foot, unable to squate , unable to jump on one foot, grade IV of muscle strength of lower extremity and slight limitation of dorsal extension of left foot. Electromyography showed multiple peripheral neurogenic lesions (motor and sensory nerve demyelination with axonal damage, more severe in lower limbs than in upper limbs). Whole exome sequencing and PCR verification indicated that the patient had a homozygous deletion in exon 4 of HK1 gene, and the variation site was located in the range of chr10:71048499-71048526, which had not been reported before, and the associated disease was peroneal muscular atrophy type 4G.
Conclusion: This study expands the number of reported cases of CMT4G and the mutation spectrum of HK1 gene, and provides reference for prenatal diagnosis and genetic counseling.
Keywords: Charcot-Marie-Tooth disease; HK1; Clinical phenotype; Gene detection; CMT4G; 0020HMSN
Introduction
Charcot-Marie-Tooth disease (CMT), also known as Hereditary Motor Sensory Neuropathy (HMSN), is a group of the most common peripheral nerve single gene genetic diseases, with a high degree of clinical and genetic heterogeneity, the prevalence rate is about 1max 2500 [1]. Its clinical features include: the average age of onset is about 12 years, progressive symmetrical muscle weakness and atrophy with sensory disorders in the distal extremities (lower limbs are more commonly involved than upper limbs), typical “crane leg-like” changes, in addition, some patients may have arched feet, spinal deformities and other manifestations [2]. The genetic mode of the disease includes autosomal dominant inheritance, autosomal recessive inheritance and x-linked inheritance, with autosomal dominant inheritance being the most common [3]. Since the discovery of the first case of CMT in 1991, with the clinical application of Whole Exon Sequencing (WES), the research of CMT has turned to the field of molecular biology. More than 100 genes have been found to be associated with CMT [4]. Due to the differences in pathological features and pathogenic genes, dozens of genotypes have been found in CMT studies, including CMT1, CMT2, CMT3, CMT4, CMT5, CMT6, CMTDI, CMTRI, CMTX and so on [5]. Of all the known pathogenic genes, the PMP22 gene that causes CMT1 is the most common, accounting for about 60.5% of all confirmed CMT cases, followed by the GBJ1 gene that can cause CMTX (about 16.7%), the MPZ gene that can cause CMT1 and CMT2 (about 9.4%), and the MFN2 gene that can cause CMT2A (about 4.4%) [6,7]. Studies have found that in patients who have been diagnosed with CMT, the positive rate of these four genes can even reach 96% [6]. It can be said that CMT4G is a rare disease among the rare diseases. CMT4G is an autosomal recessive CMT, or AR-CMT, and its pathogenic gene is HK1. A case of CMT4G caused by HK1 gene mutation is reported. The clinical data of the child is as following.
Clinical data the child, a 7-year-old girl, came to our hospital on July 10, 2021 because of “more than 7 months of unstable gait”. The child developed normally in terms of language and intelligence, lagged behind in motor development since childhood, sat steadily at more than 8 months of age, at 16 months walked independently, running, jumping and other major movements lagged behind for the same age, the left foot valgus was obvious, the left foot could not squat & the left foot jump could not be completed (Figure 1A-C). There was no numbness in the limbs or any other sensory abnormalities, and denied the history of foot trauma. While pregnant with this child, the mother was G2P2 with normal term delivery and no abnormality during pregnancy and perinatal period. Parents were healthy, had consanguineous marriage; father’s grandmother and mother’s grandmother are cousins (Figure 1D). Physical examination upon admission: the child was well oriented with normal mental status, language expression & memory. Neck supple, unremarkable cardiopulmonary & abdominal examination, equal length of both lower limbs, no obvious limitation of hip and knee movement, normal muscle tension, normal muscle strength of both the upper limbs, lower limb muscle strength of grade IV. The lateral side of the left foot is slightly swollen, the dorsal extension is slightly limited, and both arches are high. Bilateral knee reflexes were not elicited, Kirschner’s sign and Brinell’s sign were negative, and bilateral Pap’s sign was also negative. Auxiliary examination: the pelvic radiograph is normal. No obvious abnormality was found in MRI of skull and spinal cord. Electromyography and nerve conduction: the amplitudes of CMAP waves in most of the examined nerves was grossly decreased or slightly decreased, bilateral tibial nerves had conduction block, the motor nerve MCV significantly slowed down (> 3SD), the shortest latency of F wave in the right median, ulnar nerve and bilateral tibial motor nerve was prolonged, and the H-reflex latency of bilateral tibial nerves was prolonged with waveform difference (Figure 2). The amplitudes of SNAP waves of sensory nerves decreased or did not elicit, with SCV slowing down. EMG: when the tested muscle was relaxed, some of the tested muscle showed positive spontaneous potential; when the muscle contracted slightly, the MUP shape of some of the tested muscle was broadened, and the oligo-set of heavy contraction of some of the tested muscles was slightly reduced. It is suggested that the comprehensive examination results of myoelectric changes of multiple peripheral neurogenic damage (motor and sensory nerve demyelination with axonal damage, lower limbs are more severe than upper limbs) suggest peripheral neuropathy.
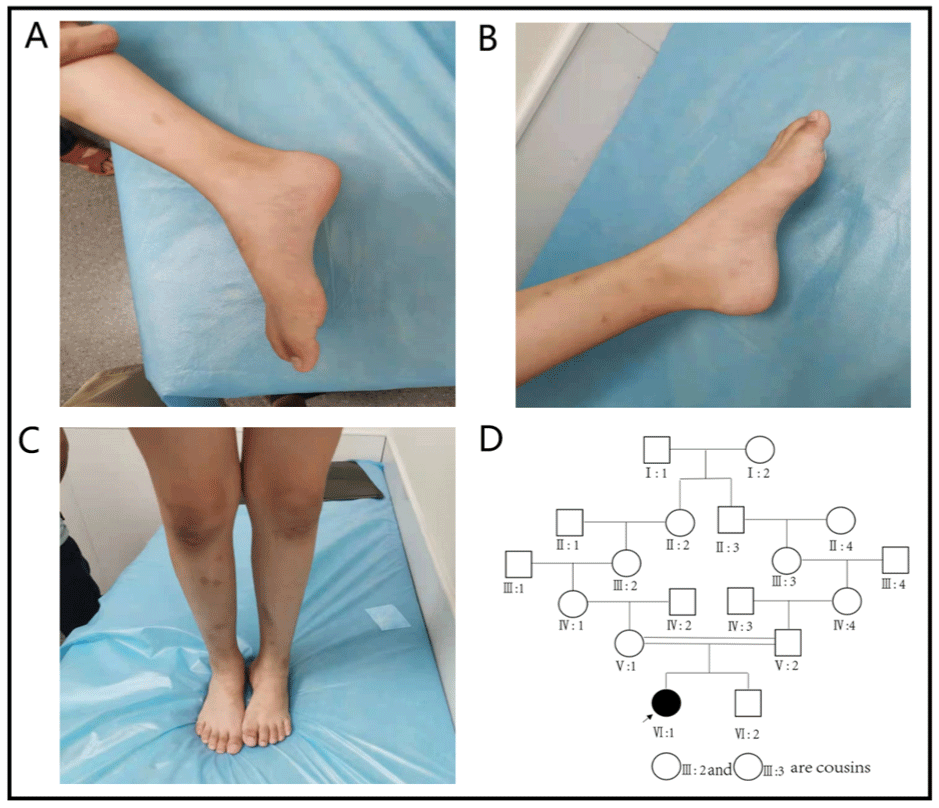
Figure 1A: No obvious deformity of the right foot; B: Obvious deformity of the high arch of the left foot; C: The deformity of the left foot is more obvious when the
child is standing; D: IV: 1 is a female proband. Father’s grandmother (III: 3) and mother’s grandmother (III: 2) are cousins, and there is a phenomenon of close
marriage in the family.
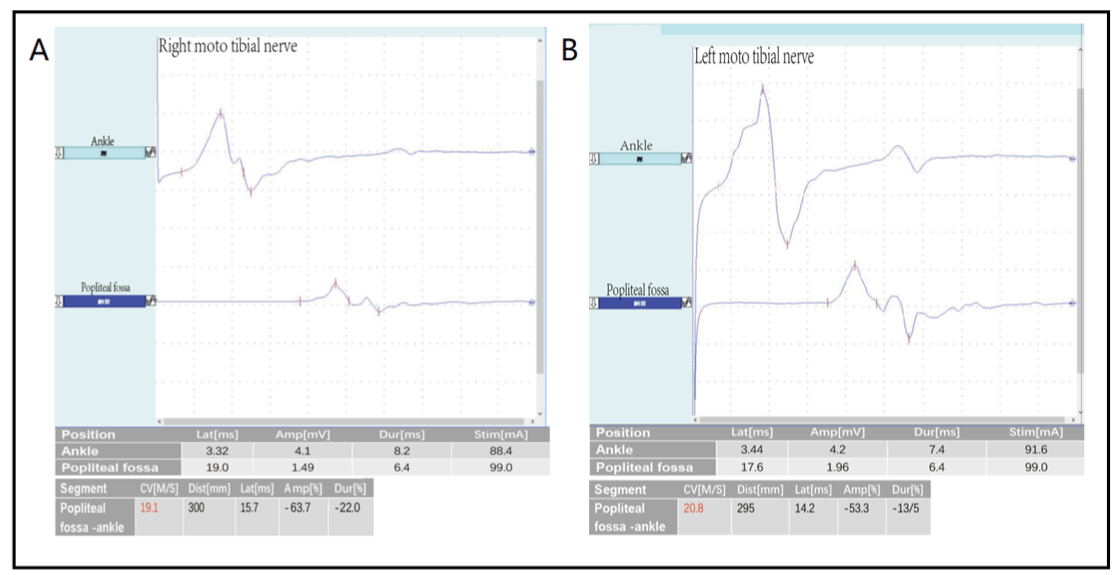
Figure 2A: The amplitudes of CMAP waves in most of the examined nerves decreased or slightly decreased, bilateral tibial nerves with conduction block, the motor
nerve MCV was significantly slowed down (> 3SD), the shortest latency of F wave in the right median, ulnar nerve and bilateral tibial motor nerve was prolonged,
and the H-reflex latency of bilateral tibial nerves was prolonged with waveform difference. A Right moto tibial nerve. B: Left moto tibial nerve.
In order to further confirm the diagnosis, with the informed consent of the guardian of the child, the peripheral blood samples of the child, his parents and brother were collected and sent to Beijing full Spectrum Medical Laboratory for gene sequencing, and the Sanger sequencing method was used for locus verification and pedigree verification. The sequencing results showed that exon 4 of HK1 gene was homozygous deletion and the mutation site was located in the range of chr10:71048499-71048526. The proband of the mutation is homozygous (Figure 3A) and conforms to the pathogenesis of Autosomal Recessive inheritance (AR). The parents of the proband are carriers respectively (Figure 3B-C). In order to further confirm the results of diagnostic sequencing, the samples of normal control, children and family members were detected by fluorescence quantitative PCR, and the exon 4 of the target gene HK1 gene was detected by using ALB gene as internal reference gene. The results showed that the ratio of copy number of exon 4 of HK1 gene in children was about 0 to that of normal controls, and the ratio of copy number of exon 4 of HK1 gene in father, mother and brother to normal controls was about 0.5 (Figure 3D). Combined with the clinical manifestations and auxiliary examination results, as well as the co-segregation of phenotypes and genotypes of the proband and his family members, the clinical diagnosis was CMT4G.
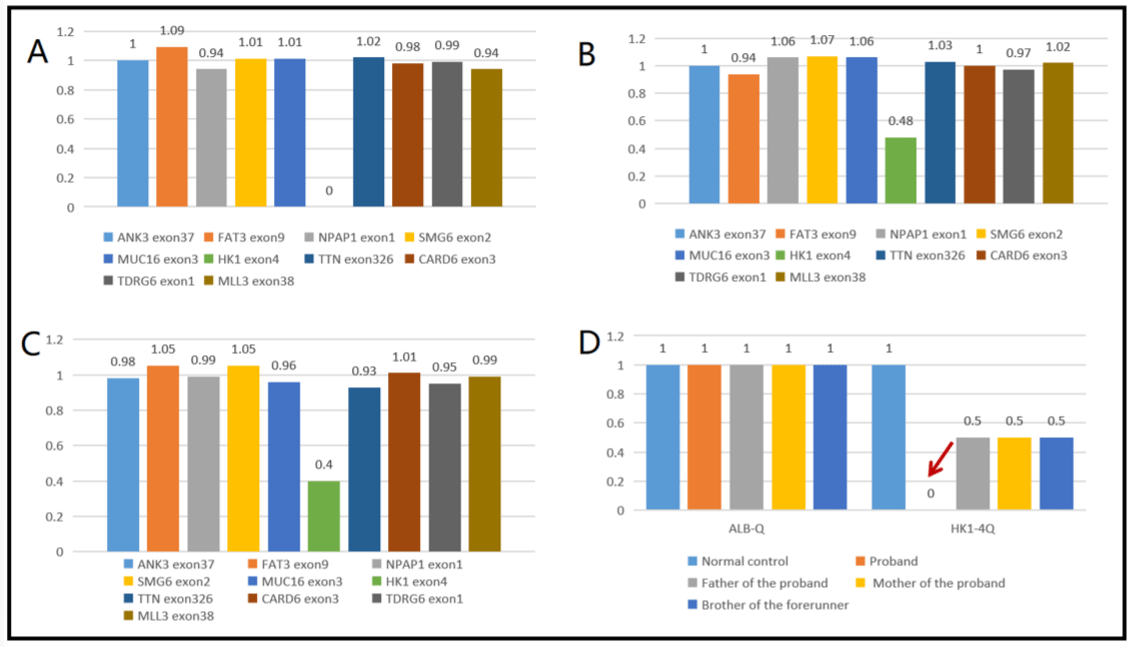
Figure 3: HK1 gene exon deletion repetition diagram. A: The proband HK14 exon homozygous deletion. B: The father of the proband lost heterozygosity in exon
HK14. C: The mother of the proband had no heterozygosity in exon HK14. D: HK1 gene exon 4 qPCR verification results; ALB-Q: internal reference gene; HK1-
4Q:HK1 gene exon 4; (note: the normal range of the ratio is 0.8-1.2). The ratio range of 1.25-1.75 is suspected to be haplorepeat, and the ratio range 1.75-2.25 is
suspected to be double repetition. The ratio range of 0.35 is less than-0.75 is suspected to be heterozygous deletion, and the ratio of 0 is homozygous deletion).
Discussion
CMT4G, also known as HMSN-Russe, is a typing of CMT. CMT4G was first found in Gypsies. In 2000, Rogers et al discovered this independent autosomal recessive disease when they studied HMSN-LOW cases in Gypsies, and located it in 10q22-23 [8]. In 2004, HMSNR was classified as CMT by ADeSandre et al for the first time, namely CMT4G [9]. The phenotype of CMT4G is more serious than that of CMT found in the past. Patients often find distal muscle weakness of lower extremities between the age of 8 and 16 years, while upper limbs are involved in different ages. With the increase of age, the symptoms gradually worsen. During the 40th to 50th years of the disease, the muscles below the knee will be severely disabled or even completely paralyzed, and in many patients, paralysis of muscles below the elbow can occur. In addition, there are a considerable number of patients with foot deformities or scoliosis. Loss of sensation is a prominent feature of the disease. In terms of nerve conduction, CMT4G showed a moderate decrease in Motor Nerve Conduction Velocity (MNCV) of the upper limb (ulnar nerve 31.9 ±7.05m/s, median nerve 32.0 ±6.8m/s), the amplitude of Compound Muscle Action Potential (CMAPs) decreased with the progression of muscle weakness and atrophy, sensory action potential loss, sural nerve biopsy specimens showed depletion of large myelinated nerve fibers. A large number of sparse myelin regenerated fiber clusters lead to an abnormal increase in the density of unmyelinated axons, and this rich regenerative activity is also the main feature of neuropathology in CMT4G [9,10]. The disease is an autosomal recessive demyelinating disease and a length-dependent axonal disease. The pathogenic gene is HK1 located in 10q22-23. HK1 encodes Hexokinase (HK). HK is not only the first catalytic enzyme in the metabolic pathway of glycolysis, but also a rate-limiting enzyme. The function of HK is to irreversibly catalyze glucose to glucose-6-phosphate, which plays a leading role in energy metabolism. HK1 is one of the four isozymes of HK, which is widely expressed in human body, mainly in red blood cells, brain, testis and other organs or cells [11]. Mutations in HK1 can cause autosomal recessive non-spherical cell hemolytic anemia, Autosomal Dominant Retinitis Pigmentosa (ADRP) and CMT4G [12]. At present, the pathogenic mechanism of HK1 mutation is not clear. Some studies have shown that HK1 may play a role in different ways: changing the regulation of myelin protein biosynthesis, disrupting axonal transport, or changing axon-Schwann cell interaction. The presence of abnormal myelinated fibers and the decrease of MNCV in sural nerve biopsies support the pathogenesis of primary myelin sheath disease. On the other hand, the obvious atrophy of proximal nerve and the retention of MNCV suggest that axonal disorder may also have an important role in this disease [9,10]. In addition, some scholars believe that HK1 mutation can lead to incomplete apoptosis and lead to CMT4G [10].
Taking “HK1 gene, peroneal muscular dystrophy” and “Hk1gene, CMT disease” as key words, we searched the articles included in China Journal full-text Database (CNKI), Wanfang data knowledge Service platform, National Biotechnology Center (NCBI) and Biomedical Literature Database (PubMed) from September to September 2021. A total of 5 related reports were found (all in English). At present, the reported HK1 mutations related to CMT4G include g.9712G > C, g.11027G > A, and c.19C > T, the first two of which appeared simultaneously in Gabrikova D et al’s 2013 report [13-15]. The homozygous deletion of exon 4 of HK1 gene was reported for the first time at home and abroad. In this case, the maternal grandmother of the father and the mother are cousins. The clinical symptoms and auxiliary examination results highly suggest peripheral neuropathy. Further whole exon sequencing showed that exon 4 of HK1 located in chr10:71048499-71048526 was missing, which has not been reported and may be a unique type in China. Fluorescence quantitative PCR analysis showed that exon 4 of HK1 was heterozygous deletion in parents and homozygous deletion in children, which conformed to the pathogenesis of Autosomal Recessive hereditary (AR) disease.
At present, 51 patients with HK1-related CMT4G have been reported, but it has not been reported in China. In the 2013 study of Sevilla T et al., 11 patients with CMT4G were found. Their clinical manifestations were as follows: distal muscle weakness of lower extremities (11amp 11), sensory loss (11pm 11), disappearance of reflex (11pm 11), foot deformity (11pm 11), scoliosis (5pm 11), MNCV < 38m/s (11pm 11). One patient found by Kanwal S et al in 2021 showed motor retardation, inability to walk alone for 15 months, abnormal gait, muscle atrophy, loss of sensation, foot deformity and disappearance of knee reflex. The clinical phenotypes of the four known mutations were not significantly different, and the genotype-phenotypic correlation was not clear. The clinical manifestation of this case is basically consistent with the above cases reported abroad. Before, CMT4G was mostly seen in the Gypsy and other ethnic minorities. Because of the high rate of inbreeding within the family and the high incidence of some hereditary diseases, this case may be the first case of CMT4G reported in China. The child and his parents have no blood disease and no family history, which limits our further tracing to the source. At present, there are few reports about CMT4G in the world, and the pathogenesis of the disease is not well understood. The emergence of this child makes up for the gap in the research of CMT4G genetic disease in China, and further expands the gene spectrum of HK1. With the progression of our work, there may be more local samples, which further suggests about the different pathogenesis of CMT4G.
The diagnosis of CMT4G still depends on full exon sequencing technology, but due to economic reasons, the high cost of test is difficult for ordinary Chinese families to afford, and the test is relatively slow, which takes 1-2 months, and long waiting often delays the disease recognition. Therefore, there is an urgent need for a relatively cheap and rapid testing as a routine test to diagnose CMT4G more easily and quickly. Recently, Matthew J et al found that an increase in GDF15 protein could be detected in the serum of patients with almost all CMT typing [16]. It can be predicted that the highly sensitive diagnostic biomarkers of CMT will be added to our clinical work in the near future.
At present, the treatment for CMT4G is very limited. The expert Guide for Children CMT in 2021[17] recommends that for children with myasthenia gravis, rehabilitation exercise is the main treatment. External fixator is feasible for children with limited range of motion. Orthopedic surgery can be sought for children with joint or spinal deformities [18,19]. In terms of targeted molecular therapy, there have been clinical trials of ascorbic acid in the treatment of CMT, but the latest research shows that ascorbic acid cannot improve the symptoms of myasthenia gravis in children with CMT [20]. In recent years, with the improvement of medical level, CMT, a rare disease, has gradually entered everyone’s field of vision. PXT3033 and ACE- 083, two orphan drugs, are popular in the treatment of CMT1. In the completed clinical trials, both of them have considerable efficacy, especially PXT3033, and can even improve nerve conduction by inhibiting the over expression of pathogenic genes [21,22]. In addition, the study of coenzyme Q10 in the treatment of CMT2A has also attracted much attention [23]. However, CMT4G is rarer than other types, and the pathogenic genes and mechanisms are also different, so the efficacy of CMT orphan drugs on CMT4G is still unknown, and we still have a long way to go for the treatment of CMT4G. In addition, many CMT patients are associated with limb pain caused by neuropathy, which can often be relieved by rehabilitation [24]. Many CMT patients suffer from mental illness, which is often neglected in our work [25]. The onset of this type of disease is early, the condition is more serious than other types, and the deterioration rate is very high, which has a great impact on their later study and life. For the patients with the disease, in addition to treatment, genetic screening should be carried out earlier, and early rehabilitation training may be the main treatment principle for a period of time in the future.
References
- Skre H, Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet. 1974; 6: 98-118.
- Chen Rong, Liang Xiuling. Clinical characteristics of fibula muscular atrophy in China. Journal of Clinical Neurology. 1996; 06: 26-29.
- McCorquodale Donald, Pucillo Evan M, Johnson Nicholas E, Management of Charcot-Marie-Tooth disease: improving long-term care with a multidisciplinary approach. J Multidiscip Healthc. 2016; 9: 7-19.
- Matilde Laurá, Menelaos Pipis, Alexander M Rossor, Mary M Reilly. Charcot- Marie-Tooth disease and related disorders: an evolving landscape. Curr Opin Neurol. 2019; 32: 641-650.
- Alexander M Rossor, James M Polke, Henry Houlden, Mary M Reilly. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013; 9: 562-71.
- Sean Ekins, Nadia K Litterman, Renée JG Arnold, Robert W Burgess, Joel S Freundlich, et al. A brief review of recent 4.4Charcot-Marie-Tooth research and priorities. F1000Res. 2015; 4: 53.
- Morena Jonathan, Gupta Anirudh, Hoyle J Chad, Charcot-Marie-Tooth: From Molecules to Therapy. Int J Mol Sci. 2019; 20.
- Rogers T, Chandler D, Angelicheva D, Thomas PK, Youl B, Tournev I, et al. A novel locus for autosomal recessive peripheral neuropathy in the EGR2 region on 10q23. American journal of human genetics. 2000; 67: 664-671.
- Sandre-Giovannoli AD, Delague V, Hamadouche T, Chaouch M, Krahn M, Boccaccio I, et al. Homozygosity mapping of autosomal recessive demyelinating Charcot-Marie-Tooth neuropathy (CMT4H) to a novel locus on chromosome 12p11.21-q13.11. Journal of Medical Genetics. 2005; 42: 260-265.
- Hantke J, Chandler D, King R, Wanders RJ, Angelicheva D, Tournev I, et al. A mutation in an alternative untranslated exon of hexokinase 1 associated with Hereditary Motor and Sensory Neuropathy – Russe (HMSNR). European Journal of Human Genetics. 2009; 17: 1606-1614.
- Koko Murakami, Hitoshi Kanno, Jakica Tancabelic, Hisaichi Fujii. Gene expression and biological significance of hexokinase in erythroid cells. Acta Haematol. 2002; 108: 204-9.
- Volkan Okur, Megan T Cho, Richard van Wijk, Brigitte van Oirschot, Jonathan Picker, et al. De novo variants in HK1 associated with neurodevelopmental abnormalities and visual impairment. Eur J Hum Genet. 2019; 27: 1081-1089.
- T Sevilla, D Martínez-Rubio, C Márquez, C Paradas, J Colomer, et al. Genetics of the Charcot-Marie-Tooth disease in the Spanish Gypsy population: the hereditary motor and sensory neuropathy-Russe in depth. Clin Genet. 2013; 83: 565-70.
- Sumaira Kanwal, Yu JIn Choi, Si On Lim, Hee Ji Choi, Jin Hee Park, et al. Novel homozygous mutations in Pakistani Kanwal Sumaira, Choi Yu JIn, Lim Si On et al. Novel homozygous mutations in Pakistani families with Charcot- Marie-Tooth disease. BMC Med Genomics. 2021; 14: 174.
- Dana Gabrikova, Martin Mistrik, Jarmila Bernasovska, Alexandra Bozikova, Regina Behulova, et al. Founder mutations in NDRG1 and HK1 genes are common causes of inherited neuropathies among Roma/Gypsies in Slovakia. J Appl Genet, 2013; 54: 455-60.
- Matthew J Jennings, Alexia Kagiava, Leen Vendredy, Emily L Spaulding, Marina Stavrou. NCAM1 and GDF15 are biomarkers of Charcot-Marie-Tooth disease in patients and mice. Brain. 2022.
- Eppie M Yiu, Paula Bray, Jonathan Baets, Steven K Baker, Nina Barisic, et al. Clinical practice guideline for the management of paediatric Charcot-Marie- Tooth disease. J Neurol Neurosurg Psychiatry. 2022.
- Reilly MM, Pareyson D, Burns J, Laurá M, Shy ME, Singh D, et al. 221st ENMC International Workshop: Foot Surgery in Charcot-Marie-Tooth disease. 10–12 June 2016, Naarden, The Netherlands. Neuromuscular Disorders. 2017; 27: 1138-1142.
- Pfeffer Glenn B, Gonzalez Tyler, Brodsky James, et al. A Consensus Statement on the Surgical Treatment of Charcot-Marie-Tooth Disease. Foot Ankle Int. 2020; 41: 870-880.
- Verhamme C, Haan RJD, Vermeulen M, Baas F, Visser MD, Schaik INV. Oral high dose ascorbic acid treatment for one year in young CMT1A patients: a randomised, double-blind, placebo-controlled phase II trial. BMC Medicine. 2009; 7: 70-70.
- Attarian S, Vallat J, Magy L, Funalot B, Gonnaud P, Lacour A. An exploratory randomised double-blind and placebo-controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot-Marie-Tooth disease type 1A. Orphanet Journal of Rare Diseases. 2014; 9.
- Pearsall R S, Davies M V, Cannell M. Follistatin-based ligand trap ACE-083 induces localized hypertrophy of skeletal muscle with functional improvement in models of neuromuscular disease. Sci Rep. 2019; 9: 11392.
- Ryoichi Takahashi, Tokuhei Ikeda, Ayumi Hamaguchi, Kazuo Iwasa, Masahito Yamada. Coenzyme Q10 therapy in hereditary motor sensory neuropathy type VI with novel mitofusin 2 mutation. Intern Med. 2012; 51: 791-3.
- Jeong Na Young, Shin Youn Ho, Jung Junyang. Neuropathic pain in hereditary peripheral neuropathy. J Exerc Rehabil. 2013; 9: 397-9.
- Cordeiro Joana L C, Marques Wilson, Hallak Jaime EC, et al. Charcot-Marie- Tooth disease, psychiatric indicators and quality of life: a systematic review. ASN Neuro. 2014; 6: 185-92.