
Case Report
Austin Pediatr. 2023; 10(1): 1082.
A Pediatric Case Report: Dysembryoplastic Neuroepithelial Tumor or Ganglioglioma?
Mezdaoui I*; Radi A; Kmari M; Hassani A; Abilkassem R; Agadr A
Department of Pediatrics, Military Hospital Mohamed V Rabat, Morocco
*Corresponding author: Mezdaoui IDepartment of Pediatrics, Military Hospital Mohamed V, Hay Riad, Rabat, 10010, Morocco. Tel: 00 212 611 22 97 50 Email: Imane.mezdaoui@gmail.com
Received: September 13, 2023 Accepted: October 19, 2023 Published: October 26, 2023
Abstract
Dysembryoplastic Neuroepithelial Tumor (DNET) is a benign glioneuronal neoplasm that most frequently affects children and young adults. It can cause chronic seizures that are medically refractory. This tumor’s cortical architecture and absence of a bulk effect or perilesional edema on radiographs are its defining features. The most typical manifestation of seizures is focal seizures with impaired awareness. Three DNET histologic subtypes have been identified. For either complicated or simple DNET types, histological detection of a distinct, particular glioneuronal component in brain tumor samples from individuals with medically refractory, chronic epilepsy serves as a diagnostic characteristic. We present one case of a child diagnosed with DNETs, who arrived at the hospital following seizures. The clinical, radiographic, histological, immunohistochemical, and molecular genetic characteristics characteristics of all three forms of DNETs as well as the differences between DNETs and gangliogliomas will be the main topics of this review.
Keywords: Child health; Epilepsy and seizures
Introduction
Dysembryoplastic Neuroepithelial Tumors (DNET) are benign tumors that are both glial and neuronal, mostly diagnosed in children and young adults [1,2], usually in the 10-14 age category. Histologically speaking, the presence of oligodendrocyte-like cells is what distinguishes them from other brain tumors [3]. DNETs usually manifest with chronic drug resistance seizures, especially focal seizures with impaired awareness [4]. We report a case of a child who presented with new seizures and found to have DNET on MRI imaging.
Case Presentation
A 4 and a half-year-old female child with no medical history and no history of seizures, presented to the hospital following a witnessed episode of generalized tonic-clonic seizures along with fever treated as a febrile seizure and was discharged from the ER.
Two months later, the child presented repeated generalized tonic-clonic seizures with loss of consciousness. The patient was then transferred to our hospital for a higher level of care.
Upon admission, she was oriented to time, place, and person. Her blood pressure and oxygen saturation were normal. The rest of her vital signs were within normal limits. The remainder of the initial examination was normal.
Routine laboratory testing, including a complete blood count and a metabolic panel were normal. The EEG recorded left temporal interictal abnormalities with activation during intermittent light stimulation. A brain Magnetic Resonance Imaging (MRI) scan was obtained, which showed a heterogeneous left parietal tumoral lesion with a double cystic and tissue composition, absence of enhancement after injection of paramagnetic contrast, thinning of the cranial vault next to the cystic lesion and absence of peri-lesional edema in favor of DNET (Figure 1).
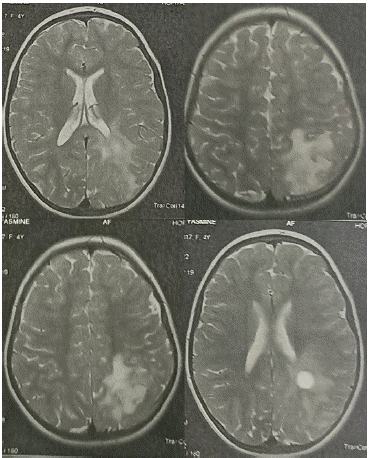
Figure 1: A brain MRI: a heterogeneous left parietal tumoral lesion with a double cystic and tissue composition.
The patient was put on carbamazepine 100 mg twice a day with remission of convulsive seizures and good progress. The patient is under frequent supervision and the surgical resection has not yet been decided.
Discussion
The incidence of DNETs is around 0.03 person-year per 100,000 with a significant peak around 10-14 years old and gets less frequent with age. 23.4% of all epilepsy-associated tumours in pediatric population are due to DNETs [1]. The sex ratio is usually in favour of males [5]. Our patient is a female child at the age of four and a half.
Clinically, DNETs are responsible for chronic drug resistant seizures [6]. They hold the second rank for the most epileptogenic brain tumour in paediatrics behind ganglioglioma [4]. Additionally, DNET may be responsible for focal seizures with impaired awareness or generalized tonic clonic forms as was the case of our patient [7]. Focal aware seizures are also present in patients [8]. Headaches could be one of DNET’s symptoms [5]. Papilledema, focal neurological deficit, behavioural changes have been mentioned in the literature as well [7].
DNETs develop mostly in the supratentorial region [9]. The most typical site of DNET is the mesial temporal lobe [8], followed by the frontal lobe and then the parieto-occipital lobe lesions [1]. While DNETs englobe all the cortex, a subcortical lesion is quite unusual [10]. The most important feature that distinguishes them is no peritumoral edema nor mass effect are found [3,6] which was the case of our patient.
According to recent research, the MRI characteristics of histologic variants of DNETs can be divided into three categories: type 1 (cystic/polycystic-like, well-delineated, strongly hypointense on T1), type 2 (nodular-like, heterogeneous signal), or type 3 (dysplastic-like, isosignal/ hypo signal T1, poor delineation, grey-white matter blurring). Simple or complex DNETs are always detected as type 1, while nonspecific DNETs are detected as type 2 or 3 (Figure 2) [4].
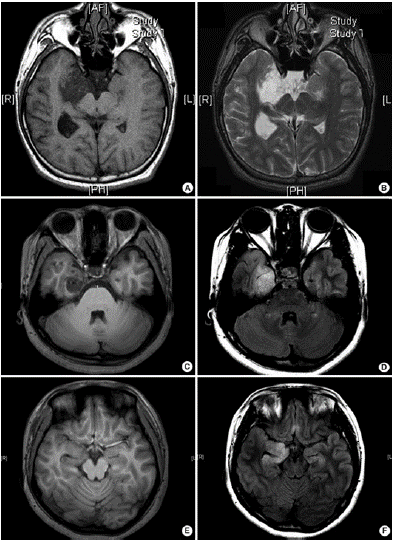
Figure 2: Three different types of magnetic resonance imaging in DNET. (A,B) Type 1 shows a well-delineated, polycystic-like tumor with strongly hypointense on T1- and hyperintense on T2-weighted images. (C,D) Type 2 shows a nodular-like, heterogeneous lesion. (E, F) Type 3 shows a poorly delineated, dysplastic-like, iso/hyposignal T1 with grey-white matter blurring [4].
The size of the lesions varies from 10 to 25mm but can possibly be larger with reported cases up to 70mm [8]. The tumors appear in the form of a solitary nodular lesion of viscous consistency [4]. A multi-nodular aspect or cystic one is frequently found [5]. Anatomopathological description of DNETs, proposed by C. Daumas-Duport in 1988, is based on the “specific glioneuronal component” characterized by the coexistence of glial cells, most often of the oligodendrocyte-like type, and of neuronal cells floating in a matrix loose interstitial [2]. Three histologic DNET types have been reported: simple, complex and non-specific [1]. We distinguish the simple forms from the complex ones where the glioneuronal component is accompanied by cortical dysplasia or glial proliferation of different nature. Nonspecific forms where the glioneuronal component is absent are also described [9]. The histological appearance is then that of low-grade gliomas and the diagnosis is confirmed on clinical and imaging elements [9] and further with immunohistochemistry [1].
Immunohistochemical techniques are often performed in addition in order to highlight antigens with diagnostic and prognostic value. In specific glio-neuronal elements, ‘floating’ neurons express neuronal markers including synaptophysin neurofilament, neuron specific enolase (NeuN), MAP2 and beta-tubulin class III [4]. The majority of oligodendroglial-like cells are strongly positive for the protein S100 and Oligo-2 but generally negative for Glial Fibrillary Acid Protein (GFAP). Rarely, OLCs may exhibit NeuN immunoexpression [5,10]. Glial nodules contain variable numbers of GFA-positive astrocytes. In the 'non-specific' form, synaptophysin is slightly less present in the lesion than in the adjacent normal cortex. Other neural markers (except MAP2) are negative [4]. CD34 expression is reported in 25–61% of cases. It is found positive in membrane of neurons, the peri-cellular stroma and in the cytoplasm of OLCs [4,8]. Ki-67 is usually low, below 1 or 2% [1]. A wide range of copy number abnormalities using whole genome sequencing were discovered, with chromosomes 5 and 7 being the most frequently altered ones [1,4].
The histogenesis of DNETs remains uncertain and remains controversial [5]. A developmental origin from the second germ layer (outer granular) has been proposed, based on the cellular mix of DNETs, their preponderance in the lobe temporalis and their association with foci of focal cortical dysplasia [3,4]. The locations in the central grey matter and on the midline suggest an origin from the subependymal germ layer. An origin from pluripotent stem cells has also been suggested due to the neuronal and glial differentiation present in OLCs. This hypothesis is supported by nestin’s and MAP2’s immunoexpression in OLCs, also implanted in neuronal and glial precursor cells during embryonic development [4].
The 3 main differential diagnoses of DNETs are represented by mostly ganglioglioma, oligodendroglioma and cortical dysplasia [8]. Gangliogliomas are more likely to have gadolinium contrast enhancement and calcifications on a CT scan [1]. Finding clusters of ganglio-like cells, granular bodies and a perivascular lymphocytic cuff is also in favour of gangliogliomas. Immunohistochemically, a CD34 positive-microtubule-associated protein 2 negative is found while DNETs result in an opposite pattern [1]. The main problem with this ambiguity between DNETs and gangliogliomas is the possible malignant transformation of the latter, which changes the whole prognosis [10]. The malignant transformation potential of DNETs is almost zero. Only one case of neoplastic degeneration of a complex shape has been described in the literature [1]. Intra-lesional hemorrhages were observed, but these were chronic bleeding, clinically occult [9].
Our patient was put on carbamazepine with good progress so far. In literature, the resection of lesions makes it possible to eliminate or control epileptic seizures [2]. The excision of these lesions therefore seems to provide a clinical benefit in the majority of cases. The radiological monitoring of resected lesions or left in place is reasonable. Imaging stability is then a key to diagnostic confirmation [2]. It is safe to say that as long as the surgical resection decision hasn’t yet been made with our patient, histological and immunohistochemical confirmation is not obtained and thus DNET diagnosis can’t be confirmed and a ganglioglioma or an oligodendroglioma are to be considered.
Conclusion
DNETs are regarded as non-recurring, benign mass lesions. They are among the most prevalent epileptogenic tumors in children. Patients with DNETs typically exhibit focal seizures with impaired awareness that are chronically drug-resistant. Lack of pathognomonic evidence is one of the difficulties in diagnosing DNETs. Our case report demonstrates a common presentation of this type of tumour although a histological and immunochemical confirmation are still to be obtained after surgery. The gold standard treatment remains complete surgical resection, which is associated with a seizure-free result in 80% to 100% of cases.
Author Statements
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of Interest
Authors report no conflict of interest our institution does not require ethical approval for reporting individual cases or case series. We confirm that all the research meets the ethical guidelines, including adherence to the legal requirements of the study country.
References
- Luzzi S, Elia A, Del Maestro M, Elbabaa SK, Carnevale S, Guerrini F, et al. Dysembryoplastic neuroepithelial tumors: what you need to know. World Neurosurg. 2019; 127: 255-65.
- Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER, Vedrenne C. Dysembryoplastic neuroepithelial tumor: A surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery. 1988; 23: 545-56.
- Chiang JCH, Harreld JH, Tanaka R, Li X, Wen J, Zhang C, et al. Septal dysembryoplastic neuroepithelial tumor: A comprehensive clinical, imaging, histopathologic, and molecular analysis. Neuro-Oncology. 2019; 21: 800-8.
- Suh YL. Dysembryoplastic neuroepithelial tumors. J Pathol Transl Med. 2015; 49: 438-49.
- Sharma MC, Jain D, Gupta A, Sarkar C, Suri V, Garg A, et al. Dysembryoplastic neuroepithelial tumor: A clinicopathological study of 32 cases. Neurosurg Rev. 2009; 32: 161-9.
- Burel-Vandenbos F, Varlet P, Lonjon M, Chanalet S, Chatel M, Michiels JF. Tumeur neuro-épithéliale dysembryoplasique à différenciation épendymaire. Ann Pathol. 2007; 27: 320-23.
- Ahluwalia R, Miles L, Hayes L, Scherer A. Pediatric septal dysembryoplastic neuroepithelial tumor (SDNT): case-based update. Childs Nerv Syst. 2020; 36: 1127-30.
- Yibirin M, De Oliveira D, Suarez I, Lombardo G, Perez C. A case of dysembryoplastic neuroepithelial tumor in an adolescent male. Cureus. 2021; 13: e13917.
- Litrico S, et al. LOCALISATION SOUS-TENTORIELLE d’une TUMEUR DYSEMBRYOPLASIQUE NEUROÉPITHÉLIALE À propos d’un cas.
- Isler C, Erturk Cetin O, Ugurlar D, Ozkara C, Comunoglu N, Kizilkilic O, et al. Dysembryoplastic neuroepithelial tumours: clinical, radiological, pathological features and outcome. Br J Neurosurg. July 4, 2018; 32: 436-41.