
Case Report
Austin Pediatr. 2025; 11(1): 1084.
A Novel Homozygous PEX5 Variant in a Newborn with Severe Zellweger Spectrum Disorder: A Case Report
Asmaa Osman1*, Lama El-Attar2, Sally Sadakaa1, Mohamed Talaat 1,3 and Wegdan Mawlana4,5
1Neonatology Department, Najran Armed Forces Hospital, Najran, Saudi Arabia
2Department of Hyman Genetics, Medical Research Institute, Alexanderia University, Egypt.
3Radiology department, Cairo University, Egypt. Head of Radiology Department, Najran Armed Forces Hospital, Saudi Arabia
4Department of Paediatrics, Faculty of Medicine, Tanta University, Egypt
5King Salman Armed Forces Hospital, Neonatology Department, Tabuk, Saudi Arabia
*Corresponding author: Asmaa Osman, Neonatology Department, Najran Armed Forces Hospital, Najran, Saudi Arabia Tel: +966556403474; Email: a_abdulla74@hotmail.com
Received: August 02, 2025 Accepted: August 25, 2025 Published: August 28, 2025
Abstract
Background: PEX gene variants play an important role in the pathogenesis of peroxisomal disorders especially Zellweger spectrum disorders (ZSDs). Clinical, biochemical screening through very long-chain fatty acids (VLCFAs) and genetic tests are valuable to reach an accurate diagnosis. The current study highlights a case of severe Zellweger spectrum in neonatal period with a novel homozygous PEX5 variant in 2babies in same Saudi family.
Case Presentation: We report a case of severe Zellweger spectrum disorder (ZSD) in a Saudi newborn 1st day of life boy (male characterized by typical clinical manifestation of hypotonia, seizures, hepatorenal insufficiency in addition to craniofacial dysmorphism, MRI Findings, and biochemical data are indicative of peroxisomal disorder. WES revealed a homozygous variant of uncertain significance (VUS) in PEX5 NM_001351132.2: c.1073T>C (p.Phe358Ser). The patient had a sibling with similar clinical, MRI and biochemical findings who died early during the neonatal period.
Conclusions: Consider ZSD in neonate with severe hypotonia, seizures and distinctive dysmorphic features in which neuroimaging finding of lissencephaly and germinolytic cysts are diagnostic clue for severe ZSD. in this report of two affected siblings born to consanguineous parents raises the suspicion of the pathogenicity of PEX5 NM_001351132.2: c.1073T>C (p.Phe358Ser) variant. It may aid in reclassifying this variant as likely pathogenic, Early diagnosis of severe ZWS could be fruitful in facilitating faster diagnosis, tailored management, and family counselling.
Keywords: Zellweger spectrum disorder; PEX5 variant; Peroxisomal disorder; Hypotonia
Introduction
Zellweger spectrum disorder (ZSD) is a rare neurodegenerative disease transmitted by autosomal recessive mode of inheritance. It represents the most severe phenotype within the peroxisomal biogenesis disorders. The worldwide prevalence of ZSD is estimated to be 1:50,000 to 1:75,000 live births [1]. ZSD is a non-curative disease with current treatment limited to supportive care. Patients with ZSD usually die in early infancy due to serious medical conditions such as respiratory failure, apnea, or infectious complications [2,3].
ZSD is characterized by reduction or absence of functional peroxisomes. Peroxisomes are membrane-bound organelles found within almost all body cells. They play a crucial role in normal lipid metabolism and cellular metabolic processes [4,5].
Functions of peroxisomes rely on proteins called peroxins, encoded by PEX (peroxisome biogenesis factor) genes essential for beta-oxidation of very long chain fatty acids (VLCFAs). Mutations in these genes disrupt organelle assembly, peroxisomal activity with subsequent elevated VLCFAs levels and multi-organ damage, particularly to the heart, eye, liver, kidney, bones, brain and nerves. Due to its progressive course and bad prognosis; early diagnosis is essential for appropriate management and genetic counselling [6,7,8].
Here we report one newborn into a close family with suspected ZWD aiming to focus on the importance of early recognition of ZSD for timely genetic counseling and proper management plan.
Case Presentation
A full-term male Saudi baby (male) born through vaginal delivery after 39 weeks of gestation; His birth weight was 2,500 g (3rd percentile), height 46 cm (between3rd and 10th percentile) and head circumference 34 cm. (50th percentile) The maternal age was 32 years old and the paternal one was 38 years old. Both parents were consanguineous (first degree relatives) and had three healthy elder siblings. The pedigree of the family shows a history of one sibling (daughter) died at neonatal period with the same clinical features and radiological presentation and that some members had unexplained hypotonia associated with brain atrophy from both the maternal and paternal side of the family with no sufficient medical reports to determine the correct diagnosis (Figure 1).
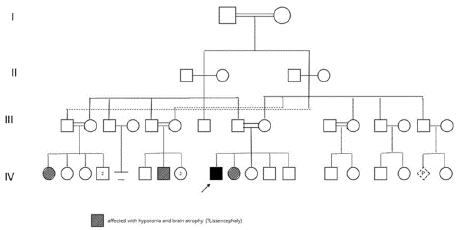
Figure 1: Pedigree of the studied case family showing positive family
history of brain atrophy and one daughter with similar clinical presentation
and radiological findings like that detected in the proband.
At birth, Delayed crying and resuscitation (PPV) for 30 second was required before developing weak cry and spontaneous breathing. The APGAR score was 5 at 1minute and 7 at 5 minutes. Full clinical and physical examination revealed severe hypotonia, weak neonatal reflexes, poor suckling, and wide anterior fontanelle communicating with posterior fontanelle. Craniofacial dysmorphism was evident in the form of high forehead, hypertelorism, depressed nasal bridge, and micrognathia. Arthrogryposis of hands and single palmar crease were seen bilaterally, bilateral talipes, and rocker-bottom feet were also present. Other findings were detected such as severe head lag, redundant neck skin, and undescended testes.
During admission in NICU, the newborn day 1 baby experienced recurrent attacks of apnea and seizures. Oral feeding was unsuccessful. So, the baby was under oxygen support, anticonvulsants, and tube feeding. A detailed workup was carried out in an attempt to reach accurate diagnosis. However, at the start laboratory investigations were normal regarding liver function tests (AST 55unit\L, ALT 25 unit /L, Albumin 32g/L), kidney function tests were as following: urea 2,7mmol\L, creatinine 47. 6umol\L. TORCH screening was negative. Regarding CBC: WBC 4,3x103 Hb 15,8 platelets 259 x103. Indirect bilirubin126 umol\L and direct bilirubin 13 umol\L while ammonia was 94 umol\L and total bilirubin 125 ummol\L.
Abdominal and pelvic ultrasound was normal at first assessment, but when repeated later on showed enlarged liver, with dilated edematous gall bladder wall and bilateral multiple renal cysts were seen. Echocardiography revealed ASD versus pfo for for follow up at 6 months.
Prominent posterior horn of lateral ventricle was evident in brain ultrasound (Figure 2). MRI brain was done in which both cerebral hemispheres showed diffuse lack of normal sulcation with multinodular surface of the cortex, congenital cortical malformation and hypoechoic cysts (Figure 3).
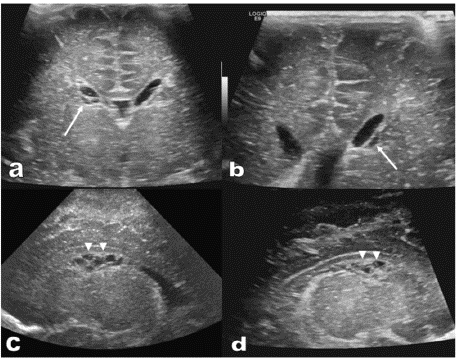
Figure 2: Brian Ultrasound Images.
(a) and (b): Trans-cranial ultrasound in coronal plane revealed cluster of
periventricular small hypoechoic cysts on both sides (white arrows).
(c) and (d): Trans-cranial ultrasound in sagittal plane demonstrated cluster
of periventricular small hypoechoic cysts centered at the caudothalamic
grooves on both sides (arrowheads).
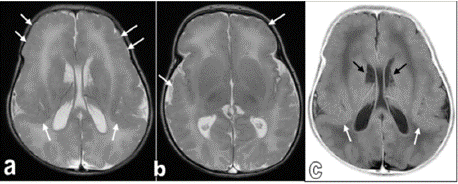
Figure 3: Brian MRI Images.
Axial T2WIs for the brain (figures a and b) and inverted axial T2WIs for the
brain (figure c). Both cerebral hemispheres showed diffuse lack of normal
sulcation with multinodular surface, more evident at the frontal and perisylvian
regions on both sides (white arrows). Findings are consistent with
congenital cortical malformation (Lissencephaly-pachygyria spectrum). In
addition, cluster of periventricular / subependymal small cysts centered
at the caudothalamic grooves on both sides (black arrows), suggestive of
germinolysis / germinolytic cysts.
However, lab investigations were repeated during follow up and revealed that liver function tests became high: ALT 175 U\L, AST:271U\L, total bilirubin 65Umol\L, direct bilirubin 54 mmo\L, with total urea became 3.1 umol\L and creatinine 23.8 umol\L. He experienced recurrent chest infection and recurrent uncontrollable seizures. Elevated levels of VLCFAs in blood sample were evident. Chromosome analysis from blood lymphocytes was done showing normal male karyotype (46, XY) with no apparent numerical or structural chromosome anomalies.
Peripheral blood sample was collected on EDTA tube and sent for whole exome sequencing (WES) by which homozygous missense variant was identified in the PEX5 gene (c.1073T>C, p.Phe358Ser).
During the hospital stay; the infant continued supportive care in the form of oxygen therapy, tube feeding, anticonvulsants, despite aggressive supportive management, the patient exhibited poor growth. When the condition of the infant becomes stable, he was referred to higher centre for proper management and genetic counselling for the family.
Discussion
Zellweger Spectrum Disorder (ZSD) is a heterogenous genetic disorder caused by a defect in one of the ZSD-PEX genes transmitted from carrier parents through autosomal recessive mode. Severe ZWD is presented early in neonatal period with unusual facial features, poor feeding, neurological and multiorgan dysfunction. Infants with severe ZSD die early during the first year of life. On the other hand, Individuals with intermediate or mild ZSD have progressive course manifested by sensorineural hearing loss, hypotonia, developmental delays, adrenal insufficiency, and liver/renal dysfunction, however intellectual function can be normal [3,9,10].
Here is a case highlights a severe clinical presentation of ZSD in an infant during the neonatal period, our case has dysmorphic features, hypotonia, difficulty in feeding and recurrent episodes of seizures which matches with severe ZSD presentation. In addition to recurrent pneumonia and chest infection. A role of peroxisomes in modulating host–pathogen interactions was suggested [11]. Fazi et al 2022 [12] hypothesized the existence of a link between ZS and humoral immunodeficiencies. However, the exact pathway and regulations of peroxisomes in relation to humoral and innate immunity is not well understood.
The associated laboratory and radiological findings in conjunction with the presence of parental consanguinity and family history of neurodegenerative disease is going classically with severe Zellweger spectrum disorders which was further supported by high level of VLCFAs as a gold standard test with the clinical picture to diagnose such cases [11], and MRI finding of lissencephaly, pachygyria and germinolytic cysts observed on MRI further support the diagnosis, as they are strongly indicative of severe ZSD when correlated with the clinical findings [13]. Alshenaifi et al 2018 [14] reported the prevalence of peroxisomal disorders among Arabs is around 1:30 000, which is much higher than the worldwide prevalence estimates [1]. The diagnosis of ZWS is based mainly on the clinical, biochemical and molecular findings in the suspected cases. Although the main biochemical marker for screening of ZWS is VLCFAs assay, the new era of molecular genetics revealed many pathogenic and likely pathogenic variants in one or more of PEX genes that are known to be involved in ZWD especially PEX1 and PEX6 gene variants [15].
Many gene variants of PEX5 gene are reported in ZWS. Based on data published before; PEX 5 gene variants were relevant in the Arabs in which peroxisomal disorders appear to be more common than in other populations (14). WES analysis of the DNA sample of the patient revealed a homozygous missense variant in the PEX5 NM_001351132.2: c.1073T>C (p.Phe358Ser). To the best of our knowledge, this novel variant has not been previously reported in the literature and not listed in CLIN Var database. According to the ACMG guidelines, this variant is classified as a variant of uncertain significance (VUS). However; based on the evident clinical manifestations of severe ZWS including dysmorphism, seizures, neurodegenerative and hepatorenal dysfunction with biochemical clues in the form of elevated VLCFAs in plasma supported by the radiological findings in the brain and kidney mentioned above in details and in addition to the family history of previous neonatal death and parental consanguinity which are further supported by the homogeneity of such gene variant in the affected proband; we can suggest that this novel PEX5 gene variant may contribute to the early neonatal mortality observed in this family and could be reclassified to likely pathogenic or pathogenic and its pathogenicity might be confirmed by further research.
Conclusion
This case emphasizes the importance of considering peroxisomal disorders in neonates with dysmorphic features, hypotonia, seizures, and characteristic brain MRI findings. Biochemical and Genetic testing plays a crucial role in early diagnosis of severe ZSD aiming to facilitate tailored management and informed family counseling.
Abbreviations
ZDS: Zellweger spectrum disorder; PEX: peroxisome biogenesis factor; VLCFAs: very long chain fatty acids; PPV: positive pressure ventilation; NICU: neonatal intensive care unit; CBC: complete blood count; ALT: alanine transaminase; WES: whole exome; ZWD: Zellweger disease; MRI: magnetic resonance imaging; ZWS: Zellweger syndrome; VUS: variant of unknown significant.
Declarations
Ethics Approval and Consent to Participate
Ethics approval was not required for this case report
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report
Availability of Data and Materials
Available in Armed Forces Hospitals Southern Region, Najran
Competing interests
All financial and non-financial competing interests must be declared in this section.
Authors' Contributions
Dr. Asmaa Abdulla Mohamed Osman: Recognize case that is unusual, has valuable clinical insights, Critical analysing case highlights its unique features, Draft manuscript, Respond to peer review
Dr. Lama M. El -Attar: Drafting manuscript, clearly explaining the significance of the case from genetic side of view, Review literature, place the case in context and compare it to similar cases or known knowledge, review manuscript, respond to peer review
Dr. Sally Ibrahim Hafez Sadaka: Collecting the data and accurate clinical information, ensuring data has history, physical examination diagnostic tests interventions, and outcomes
Dr. Mohamed Talaat Ali Abdallatif: Draft manuscript, Explain radiological importance of baby findings, Collaboration with coauthors
Dr. Wegdan Mawlana: Ensure ethical standards, respond to peer review, making revisions to improve the manuscript, Promotion knowledge sharing.
Acknowledgements
Authors thank parents for their co-operation.
References
- Lee PR, Raymond GV. Child neurology: Zellweger syndrome. Neurology. 2013; 80: e207-e210.
- Poll-The BT, Gootjes J, Duran M, de Klerk JBC, Maillette de Buy Wenniger- Prick LJ, et al. Peroxisome Biogenesis Disorders with Prolonged Survival: Phenotypic Expression in a Cohort of 31 Patients. Am. J. Med. Genet. 2004; 126A: 333–338.
- Braverman NE, Raymond GV, Rizzo WB, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016; 117: 313- 321.
- Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006; 75: 295-332.
- Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006; 1763: 1733- 1748.
- Moser AE, Singh I, Brown FR 3rd, et al. The cerebrohepatorenal (Zellweger) syndrome. Increased levels and impaired degradation of very-long-chain fatty acids and their use in prenatal diagnosis. N Engl J Med. 1984; 310: 1141- 1146.
- Grayer J. Recognition of Zellweger syndrome in infancy. Adv Neonatal Care. 2005; 5: 5-13.
- Levesque S, Morin C, Guay SP, et al. A founder mutation in the PEX6 gene is responsible for increased incidence of Zellweger syndrome in a French Canadian population. BMC Med Genet. 2012; 13: 72.
- Rafique M, Zia S, Rana MN, Mostafa OA. Zellweger syndrome - a lethal peroxisome biogenesis disorder. J Pediatr Endocrinol Metab. 2013; 26: 377- 379.
- Bose M, Yergeau C, D’Souza Y, et al. Characterization of Severity in Zellweger Spectrum Disorder by Clinical Findings: A Scoping Review, Meta-Analysis and Medical Chart Review. Cells. 2022; 11: 1891.
- Di Cara F. Peroxisomes in host defense. PLoS Pathog. 2020; 16: e1008636.
- Fazi C, Lodi L, Magi L, et al. Case Report: Zellweger Syndrome and Humoral Immunodeficiency: The Relevance of Newborn Screening for Primary Immunodeficiency. Front Pediatr. 2022; 10: 852943.
- Weller S, Rosewich H, Gärtner J. Cerebral MRI as a valuable diagnostic tool in Zellweger spectrum patients. J Inherit Metab Dis. 2008; 31: 270- 280.
- Alshenaifi J, Ewida N, Anazi S, et al. The many faces of peroxisomal disorders: Lessons from a large Arab cohort. Clin Genet. 2019; 95: 310-319.
- Ebberink MS, Mooijer PAW, Gootjes J, Koster J, Wanders RJA, Waterham HR. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat. 2011; 32: 59–69.