
Case Report
Austin J Pediatr. 2015; 2(2): 1021.
Critical Pulmonary Stenosis with Progressive Hypertrophic Cardiomyopathy
Bennett Samuel, Candida Pinto, Joseph Vettukattil*
Congenital Heart Center, Helen DeVos Children’s Hospital of Spectrum Health, USA
*Corresponding author: Joseph Vettukattil, Co- Director, Congenital Heart Center and Division Chief, Pediatric Cardiology, Helen DeVos Children’s Hospital of Spectrum Health, 100 Michigan NE (MC273), Grand Rapids, Michigan 49503, USA
Received: May 28, 2015; Accepted: September 15, 2015; Published: October 06, 2015
Abstract
Carnitine acylcarnitine translocase (CACT) deficiency and carnitine palmitoyltransferase type II (CPT II) deficiency are severe, lethal mitochondrial fatty acid oxidation disorders (FODs). These FODs are associated with cardiomyopathy, life-threatening arrhythmias, severe liver dysfunction, hypoketotic hypoglycemia, moderate-to-severe hypotonia and failure to thrive. As fetal metabolism is carried out by maternal circulation the clinical symptoms of metabolic deficiency are only observed after 24 to 36 hours of age when the neonate beings to metabolize on its own. Structural heart defects such as pulmonary valve stenosis are not known to be associated with FODs. We describe a term neonate with critical pulmonary stenosis relieved by balloon valvuloplasty that developed progressive fatal left ventricular hypertrophy within five days. Neonates presenting with critical pulmonary valve stenosis who develop unexplained ventricular hypertrophy should be tested for metabolic disorders for appropriate management of these patients.
Introduction
The neonatal form of fatty acid oxidation disorder (FOD) is associated with high mortality. The core conditions of FODs include carnitine uptake defect/carnitine transport defect, medium-chain acyl-coenzyme A (CoA) dehydrogenase deficiency, long-chain acyl- CoA dehydrogenase deficiency, trifunctional protein deficiency and very long chain acyl-CoA dehydrogenase deficiency. Carnitine palmitoyltransferase type I deficiency, carnitine palmitoyltransferase type II (CPT II) deficiency, carnitine-acylcarnitine translocase (CACT) deficiency, glutaric academia type II, medium-chain ketoacyl- CoA thiolase deficiency, medium-/short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency and short-chain acyl-CoA dehydrogenase deficiency are known secondary conditions. The common signs and symptoms include cardiomyopathy, life-threatening arrhythmias, severe liver dysfunction, hepatic encephalopathy, hypoketotic hypoglycemia, moderate-to-sever hypotonia, lactic acidosis, and failure to thrive [1]. However, structural defects such as pulmonary valve stenosis are not known to be associated with FODs in the literature. We describe critical pulmonary stenosis and rapidly progressing fatal cardiomyopathy in a term neonate with elevated palmitoylcarnitine (C16), hexadecenoylcarnitine (C16.1) and stearoylcarnitine (C18) suggesting a diagnosis of either CACTdeficiency or CPT II deficiency.
Case Report
A term 3.29 kg male neonate (63rd percentile) of African descent was diagnosed with critical pulmonary stenosis at birth and developed rapidly progressing hypertrophic cardiomyopathy. Parental consanguinity was unknown. His APGAR scores were within normal limits. The newborn’s length was 51 cm (82nd percentile) and head circumference was 33 cm (31st percentile). There was no reported exposure to alcohol, tobacco, or illicit drugs during fetal development, but documentation of prenatal care was unavailable for review. Human immunodeficiency virus and hepatitis screen were negative.
He was not distressed at rest and had normal clinical findings. However, shortly after birth, he was noted to have a loud crescendo systolic murmur grade II-III/VI in the 2nd left intercostal space. An echocardiography confirmed critical pulmonary valve stenosis with peak gradient of 71 mmHg and mean gradient of 33 mmHg across the valve. Dynamic right ventricle outflow tract obstruction was defined. Pulmonary valve morphology was not able to be clearly determined on echocardiogram. The patient was stabilized with prostaglandin E2 and a cardiac catheterization and balloon valvuloplasty was scheduled for day 3 after birth.
On echocardiogram and angiogram, the pulmonary valve annulus measured between 7.5-7.7 mm. A 9x20 mm Tyshak balloon dilatation catheter (Braun Interventional Systems, Inc., Bethlehem, PA, USA) was used for valvuloplasty using standard techniques. Following the procedure, the residual valvar gradient was 18 mmHg (49 to 31 mmHg). There was significant residual dynamic subpulmonary gradient of approximately 50 mmHg. He received an increasing dose of propranolol to manage the dynamic sub pulmonary stenosis. Other observations included mildly hypoplastic pulmonary valve annulus, normal tricuspid value annulus, right ventricle forming with severe right ventricular hypertrophy, patent foramen ovale with a small to moderate right-to-left shunt, small patent ductus arteriosus, small left-to-right ductal shunt and normal left ventricular dimensions and systolic function.
After several episodes of hypoketotic hypoglycemia and abnormal labs such as hyper-ammonemia (259.1 μmol/L), elevated creatinine kinase (4,024 U/L), aspartate transaminase (420 IU/L) and alanine transaminase (116 IU/L), the infant’s condition deteriorated rapidly on day 5. Despite oxygenation, it was difficult to maintain saturations above 80%. The neonate was placed on continuous positive airway pressure with no improvement and required intubation. The left ventricular posterior wall thickness in diastole rapidly progressed from 0.40 cm on day 0 to 0.81 cm on day 5 (Table 1; Figure 1A and 1B). The newborn screening reported elevated levels of C16 (13.84 μmol/L), C16.1 (2.51 μmol/L) and C18 (1.59 μmol/L). The infant died of severe ventricular diastolic dysfunction with progressive pulmonary edema and low cardiac output.
![]()
RV anterior wall thickness in systole (mm)
RV anterior wall thickness in diastole (mm)
Shunting
LVPWd (cm)
LV mass (g)
IVSd (cm)
Day 0
6.1
4.1
Right-to-left
0.4
14.9
0.43
Day 4
7.7
6.1
Bidirectional
0.46
14.1
0.58
Day 5
12
9.2
Left-to-right
0.81
19.3
0.75
Note. IVSd : Interventricular septum in diastole; LVPWd – left ventricular posterior wall in diastole; LV – left ventricle; RV – right ventricle.
Table 1: Comparison of echocardiogram measurements on day 0, day 4, and day 5.
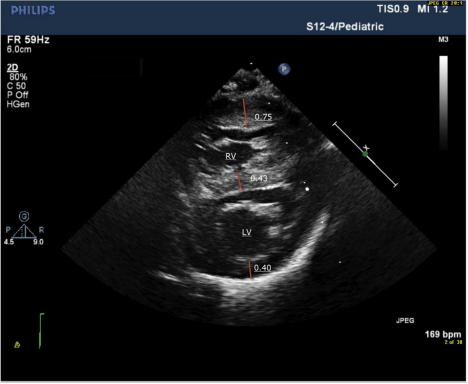
Figure 1A: Short axis echocardiogram on day 0 showing severe right
ventricular hypertrophy with normal left ventricular thickness.
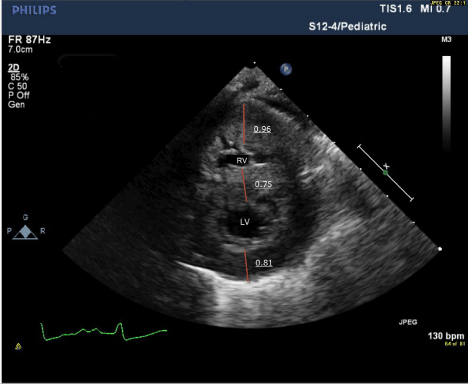
Figure 1B: Short axis echocardiogram on day 5 showing progression of left
ventricular hypertrophy.
Discussion
Mutations in the SLC25A20 gene causes CACT deficiency and mutations in CPT2 gene causes CPT II deficiency. Although rapid decline in condition due to lethal neonatal form of CACT and CPT II are described in the literature, it is not known to be associated with structural defects such as pulmonary valve stenosis. The neonate was diagnosed with severe pulmonary stenosis at birth, which was managed with balloon valvuloplasty. The right ventricular hypertrophy was appropriate for the amount of stenosis noted and the left ventricle size was normal. The rapid progression of left ventricular hypertrophy suggests a degree of metabolic imbalances leading to deposition of metabolites in the myocardium causing both hypertrophy and myocardial dysfunction.
The manifestation of FODs is usually seen 24-36 hours after birth as the child begins its own metabolism [1]. These disorders are difficult to identify in utero as metabolism takes place in the maternal circulation and supports the fetus. The usual presenting symptoms are nonketotic hypoglycemia, hepatomegaly, elevated liver enzymes, hyperammonemia, cardiomyopathy and neurological abnormalities [2]. The therapy for infantile form of FOD is a diet rich in carbohydrates, low fat, and supplemented with carnitine. However, the fatal neonatal form of CACT and CPT II deficiency responds poorly to treatment [3].
The diagnosis of CACT and CPT II is suspected with abnormal newborn screening results such as low free carnitine and elevated C16-18 levels, which is further confirmed by genetic tests. Although neonatal screening allows for early diagnosis of FODs, the severe neonatal form of CACT and CPT II deficiency may progress rapidly to irreversible multiple organ dysfunctions even before the results become available. The two FODs are not distinguishable and further genetic testing with enzyme assay in fibroblasts or DNA analysis confirms the diagnosis [3]. Yang, Mallory, Roe and colleagues describe prenatal diagnosis using enzyme assay to identify CACT and CPT II deficiency in a mother with previous history of a child with FOD [4]. As observed in our case, echocardiography from day 1 or 2 of life in patients with FODs does not show typical findings of metabolic cardiomyopathy. Therefore, repeat serial echocardiographies are mandatory.
The timely diagnosis and treatment of patients with FODs can result in positive outcomes. Unfortunately, the severe neonatal forms of FODs have poor prognosis and high mortality rates. Although cardiomyopathy is expected in CACT and CPT II deficiency, structural defects such as pulmonary valve stenosis have not been associated with FODs in the literature. The rapidity of progression of hypertrophic cardiomyopathy would have been unlikely to be detected if our patient did not initially present with critical pulmonary stenosis. Neonates presenting with critical pulmonary valve stenosis who develop unexplained ventricular hypertrophy should be tested for metabolic disorders. Repeat serial echocardiograms are necessary to diagnose and appropriately manage the condition.
References
- Saudubray JM, Martin D, de Lonlay P, Touati G, Poggi-Travert F, Bonnet D, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999; 22: 488-502.
- Anderson S. Brooks SS when the usual symptoms become an unusual diagnosis: A case report of trifunctional protein complex. Neonatal Netw. 2013; 32: 262-273.
- Longo N, Amat di San Filippo C and Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet 142C. 2006: 77-85.
- Yang BZ, Mallory JM, Roe DS, Brivet M, Strobel GD, Jones KM, et al. Carnitine/acylcarnitine translocase deficiency (neonatal phenotype): Successful prenatal and postmortem diagnosis associated with a novel mutation in a single family. Mol Genet Metab. 2001; 73: 64-70.