
Special Article – Pediatric Case Reports
Austin Pediatr. 2016; 3(3): 1035.
An Uncommon Presentation of Bohring-Opitz Syndrome in a 2 Week-Old Newborn Female
Gonzalez de Alba CE1*, Berganza FM1, Sawhney R2 and Ojadi V3
1Driscoll Children’s Hospital, Department of Pediatrics, Corpus Christi, Texas, USA
2Texas College of Osteopathic Medicine, University of North Texas Health Science Center, Fort Worth, Texas, USA
3Driscoll Children’s Hospital, Department of Neonatology, Corpus Christi, Texas, USA
*Corresponding author: Cesar E Gonzalez de Alba, Department of Medical Education, Driscoll Children’s Hospital, 3533 S Alameda St, Corpus Christi, Texas, USA
Received: July 13, 2016; Accepted: August 02, 2016; Published: August 04, 2016
Abstract
Bohring-Opitz Syndrome (BOS) is a rare genetic syndrome that was first described by Bohring et al in 1999. He reported 2 cases of infants with Opitz trigonocephaly (C)-like syndrome as well as 2 similar published cases previously assumed to have C syndrome. BOS has been associated with distinct facial characteristics such as prominent metopic suture, exophthalmos, hypertelorism, cleft lip and palate, microcephaly, intrauterine growth retardation, feeding difficulties, flexion deformities of the upper limbs (referred to as ‘BOS posture’), among other anomalies. We present the case of a 2 week old baby girl with BOS who was transferred to our facility with congestive heart failure (CHF) secondary to a large anterior VSD, ASD, with elevated transaminases. To our knowledge, this is the first case of Bohring-Opitz Syndrome ever reported with such clinical presentation.
Keywords: ASXL1; Bohring-Opitz Syndrome; Congenital heart disease
Abbreviations
ASXL1; ASD: Atrial Septal Defect; BOS: Bohring-Opitz Syndrome; CHF: Congestive Heart Failure; FOC: Fronto-Occipital Circumference, IUGR: Intra Uterine Growth Retardation, VSD: Ventricular Septal Defect
Case Presentation
The infant was a 2½-week-old female, seen in referral at Driscoll Children’s neonatal intensive care unit for evaluation of Atrial and ventricular septal defects (ASD and VSD respectively) with congestive heart failure (CHF), intrauterine growth retardation (IUGR) and dysmorphic features. Family history was not recorded. Mother was a 25-year-old primigravida and prenatal care was provided in Mexico. The infant was born at 36 weeks gestation by cesarean section for pregnancy induced hypertension at an outside facility. There were no complications at birth. Apgar scores were 3/8. Birth weight was 1560g, length 39cm. Microarray was normal. With development of CHF, she was transferred to Driscoll Children’s NICU.
Patient’s admission length was 41cm (z= -4), weight 1552gm (z= -4) and FOC of 29.6cm (z= -3). The head was trigonocephalic with movement of the metopic suture at the fontanel but with ridging along the lower ¾. The anterior fontanel was 1½ x 1½ cm. The posterior whorl was not seen and there was a hirsute forehead with bifrontal upsweeps. The palpebral fissures were slightly up-sloped and 12 mm OU. Inner canthal distance was 17mm. The eyebrows blended into lateral forehead hirsutism. The corneas were clear with no apparent cataract. There was hollowing above the supra orbital ridges. The nasal bridge was normal with the tip of 20mm with normal alae. There was limited opening of the mouth with a high-arched palate and wide alveolar ridges. The jaw and neck were normal. The right ear was 36 mm long with a broad antihelix. The left ear was 32mm long with decreased cartilage and slight over folding of the superior helix. It was placed at the outer canthal-inion line. The chest was symmetric with no bony abnormalities. Circumference was 26cm, inter-nipple distance was increased at 7.4cm and sternal length of 4.7cm. There was increased RV impulse, normal LV impulse, single S1, single S2, 2-3/6 systolic regurgitant murmur at URSB; no gallop, no rubs. The abdomen appearance was normal, with the liver edge down 4cm and the umbilicus normally placed. The genitalia were that of a normal preterm female. The anus was anteriorly placed with a pit or fistula in the perineal space. The upper extremities were proportionate with full extension and supination. The palmar creases, thumbs and nails were normal. The fifth fingers were short with a single interphalangeal crease and there was wrist flexion and mild ulnar drift of both hands. There were no significant pigmentary or vascular changes of the skin. Muscle tone was increased in the arms and legs. A genetics consultation was obtained, and it was determined that the patient’s physical findings best fitted Bohring-Opitz Syndrome (Figures 1 and 2).
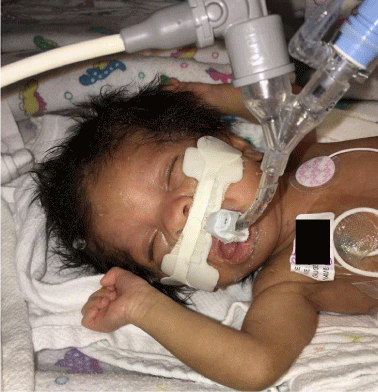
Figure 1: Characteristic facial phenotype of BOS with prominent metopic
suture and trigonocephaly.
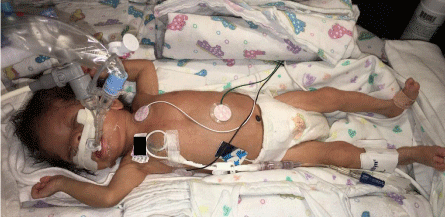
Figure 2: Full body of the patient is depicted. Notice the increased muscle
tone in arms with wrist flexion and mild ulnar drift of both hands (better
appreciated on the right wrist).
Patient’s admission echocardiogram showed a 5mm low secundum ASD with left to right shunt and a large (6mm) membranous VSD with mild anterior malalignment with bidirectional flow. Right atrium was moderately dilated while the right ventricle was normal in size with mild hypertrophy and qualitatively normal systolic function. Hepatic veins, right and pulmonary arteries were all dilated. Admission lab studies showed elevated liver enzymes, ammonia, BUN and lactate. Baby gram showed 13 pairs of ribs. Head sonogram showed an absent corpus callosum. A brain magnetic resonance imaging study confirmed the agenesis of the corpus callosum as well as suspected focal stenosis at the craniocervical junction. The pituitary gland was not well visualized, however a blood hormonal profile was negative for any deficiency.
A complete ASXL1 gene sequence analysis was sent to Fulgent labs (Temple City, CA). No clinically significant mutations were identified. Prepared DNA libraries were sequenced using Next Generation Sequencing technology.
Discussion
Bohring-Opitz Syndrome (BOS) is a rare genetic syndrome that was first described by Bohring et al in 1999. He reported 2 cases of infants with Opitztrigonocephaly (C)-like syndrome as well as 2 similar published cases previously assumed to have C syndrome [1-2]. To our knowledge, there are only 52 published cases of BOS in the literature today [3-5]. The oldest known BOS patient was last described in 2011 when he was 24 years of age, but a literature review failed to reveal any additional information about his current status [6]. He represents a minority however; as BOS has a 40% infant mortality rate and many patients die before 5 years of age [7]. Mortality is usually due to recurrent infections, obstructive apnea, unexplained bradycardia, or respiratory distress/failure [5-7].
The mechanism and inheritance pattern of BOS remain unclear, however de novo heterozygous frame shift or nonsense mutations in ASXL1 gene have been identified in ten of cases of BOS by different authors [3,6,8]. The ASXL1 gene has been associated with both activation and silencing of HOX genes, which are involved in body segment formation, chromatin remodeling, as well as having oncogene properties [9,10]. Interestingly, not all patients with BOS have abnormalities in their ASXL1 gene, suggesting the possibility of multiple etiologies [6,7]. Mutations of the ASXL3 gene, which is part of the same gene family as ASXL1, have been described in five distinct cases [11,12]. While these patients had similar characteristics as those found in patients with BOS, they did not fully exhibit the specific characteristics defined for BOS [3].
Diagnosis of BOS usually occurs during the neonatal period, based on the patient’s clinical picture. Hastings, et al [7] proposed a set of diagnostic criteria in which 7 out of 10 features must be present: typical facial appearance (trigonocephaly/prominent metopic ridge, retrognathia, prominent eyes with hypoplastic supraorbital ridges, upslanting palpebral fissures, depressed nasal bridge, anteverted nares, low-set and posteriorly rotated ears, palatal abnormalities and broad alveolar ridges, flammeus nevus, low anterior hairline), microcephaly, IUGR and short stature, joint abnormalities, abnormal tone, severe/ profound developmental delay, susceptibility to infections, feeding difficulties, and high infant mortality. Other characteristics described in other reports include hirsutism, exophthalmos, and low frontal and temporal hairline. Systemic manifestations have also been described, including gastrointestinal, ophthalmologic, cerebral and cardiac anomalies. Of the latter, the cardiac defects specifically described in the literature include: pulmonary hypertension, biventricular hypertrophy, patent ductus arteriosus (PDA), patent foramen ovale (PFO), dysplastic pulmonary valve with mild stenosis, atrial septal defect (ASD), and perimembranous ventricular septal defect (VSD [2,4-7]. Given the small number of reported cases of BOS, it is difficult to establish the significance of other abnormalities as unique to BOS or as manifestations of concomitant conditions.
Even though we did not find a mutation in the ASXL1 gene sequencing in our patient, her clinical presentation was characteristic for BOS, meeting more than 7 of the “classic” criteria, including IUGR/short stature, microcephaly, trigonocephaly with movement of the metopic suture at the fontanel, upslanting palpebral fissures, a high-arched palate with wide alveolar ridges, low anterior hairline with hirsutism, classic “BOS posture” with wrist flexion and mild ulnar drift of both hands, abnormal tone, feeding difficulties as well as systemic manifestations in the gastrointestinal, brain and cardiac systems.
Treatment of BOS is supportive and often includes a gastrostomy tube (G-tube) for feedings and mechanical ventilation in the neonatal period. As patients survive through early childhood, problems such as feeding difficulties and recurrent infections become less significant. Despite early intervention, profound intellectual delay tends to persist, although there is case by case variability in severity [7]. Children who survive into their teenage years and adulthood still face significant morbidity despite decreased mortality. Our patient was scheduled for pulmonary artery banding as a palliative measure to help reduce the flow to her pulmonary vascular circuit.
References
- Oberklaid F, Danks DM. The Opitz trigonocephaly syndrome: A case report. Am J Dis Child. 1975; 129: 1348-1349.
- Bohring A, Oudesluijs GG, Grange DK, Zampino G, Thierry P. New cases of Bohring-Opitz syndrome, update, and critical review of the literature. Am J Med Genet A. 2006; 140: 1257-1263.
- Dangiolo SB, Wilson A, Jobanputra V, Anyane-Yeboa K. Bohring-Opitz syndrome (BOS) with a new ASXL1 pathogenic variant: Review of the most prevalent molecular and phenotypic features of the syndrome. Am J Med Genet A. 2015; 167: 3161-3166.
- Russell B, Johnston JJ, Biesecker LG, Kramer N, Pickart A. Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am J Med Genet A. 2015; 167: 2122-2131.
- Bohring A, Silengo M, Lerone M, Superneau DW, Spaich C, Braddock SR, et al. Severe end of Opitztrigonocephaly (C) syndrome or new syndrome? Am J Med Genet. 1999; 85: 438-446.
- Hoischen A, van Bon BW, Rodríguez-Santiago B, Gilissen C. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011; 43: 729-731.
- Hastings R, Cobben JM, Gillessen-Kaesbach G, Goodship J, Hove H. Bohring-Opitz (Oberklaid-Danks) syndrome: clinical study, review of the literature, and discussion of possible pathogenesis. Eur J Hum Genet. 2011; 19: 513-519.
- Magini P, Della Monica M, Uzielli ML, Mongelli P, Scarselli G, Gambineri E, et al. Two novel patients with Bohring-Opitz syndrome caused by de novo ASXL1 mutations. Am J Med Genet A. 2012; 158A: 917-921.
- Avila M, Kirchhoff M, Marle N, Hove HD, Chouchane M, Thauvin-Robinet C, et al. Delineation of a new chromosome 20q11.2 duplication syndrome including the ASXL1 gene. Am J Med Genet A. 2013; 161A: 1594-1598.
- Guo YB, Shao YM, Chen J, Xu SB, Zhang XD, Wang MR, et al. Effect of overexpression of HOX genes on its invasive tendency in cerebral glioma. Oncol Lett. 2016; 11: 75-80.
- Dinwiddie DL, Soden SE, Saunders CJ, Miller NA, Farrow EG, Smith LD, et al. De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. 2013; 6: 32.
- Bainbridge MN, Hu H, Muzny DM, Musante L, Lupski JR, Graham BH, et al. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med. 2013; 5: 11.