
Case Report
Austin Pediatr. 2017; 4(1): 1051.
Refractory Hypoglycemia in a 2-Month Old Female
Brunson C¹ and Chadha T2,3*
¹Department of Pediatrics, Children’s Hospital of Philadelphia, Philadelphia, USA
²Department of Pediatrics, Wolfson Children’s Hospital, Jacksonville, USA
³Department of Pediatrics, University of Florida College of Medicine, Jacksonville, USA
*Corresponding author: Tanya Chadha, Department of Pediatrics, Wolfson Children’s Hospital, Jacksonville, USA
Received: April 03, 2017; Accepted: April 18, 2017; Published: April 25, 2017
Abstract
In this case report, we describe a rare cause of persistent hypoglycemia in a young infant. The case briefly highlights for the pediatric trainee the broad differential diagnosis for hypoglycemia, a commonly encountered laboratory abnormality in the young infant population and the importance of history and physical exam in arriving at a final diagnosis.
Keywords: Hypoglycemia; Infants; Growth hormone; Hypopituitarism; Sepsis
Case Presentation
A 2-month old full term Hispanic female presented to the emergency room with an initial complaint of no bowel movement for the past 24 hours. On further questioning the parents also state that the patient had a decrease in the amount of feeding overnight into the morning of presentation. Her parents deny any fever, irritability, vomiting or diarrhea and don’t endorse any toxic ingestion or recent trauma. The patient’s past medical history is significant for neonatal intensive care unit admission at birth for observation secondary to apneic episodes and hypoglycemia briefly requiring dextrose containing intravenous fluids to normalize her blood glucose levels.
On presentation to the emergency room she was found to be lethargic with no other significant findings on physical exam. Of note, she was found to have dysmorphic facies. Vital signs were as follows: temperature 97 degrees Fahrenheit, heart rate 139 beats per minute, respiratory rate 22 breaths per minute, blood pressure 103/48 mmHg and oxygen saturation 100% on room air. The patient’s lethargy prompted a dextrose stick which was <35mg/dL for which she received an IV dextrose bolus with transient improvement in her mental status. However, she continued to have episodes of lethargy associated with significant hypoglycemia that required IV dextrose for correction. She was transferred to the pediatric intensive care unit for further evaluation.
The patient’s initial laboratory evaluation was significant for glucose level of 20mg/dl with otherwise normal serum electrolytes and normal liver function studies. With low suspicion of ingestion based on history, sepsis was considered the likely source of hypoglycemia and thus a work-up was completed and broad spectrum antibiotics were started. The white blood cell count was normal and urinalysis was negative for leukocyte esterase, nitrites and ketones. Blood, urine and cerebrospinal fluid cultures were all subsequently found to be negative. With ongoing hypoglycemia requiring significant intravenous dextrose supplementation, development of abnormal movements concerning for seizures and dysmorphic faces noted upon presentation, multiple subspecialists were consulted for further evaluation and a number of laboratory and imaging diagnostics were pursued. Video electroencephalography was normal. A magnetic resonance imaging study of the patient’s brain revealed an ectopic posterior pituitary gland. A chromosomal evaluation was found to be abnormal. Laboratory investigation demonstrated a low growth hormone level of 0.87ng/mL (normal 5-40ng/mL), a normal insulinlike growth factor-1 level of 25ng/ml (normal 17-185 ng/mL), a low free thyroxine level of 0.73ng/dL (normal 0.9 -1.9ng/dL) and a low cortisol level of 3.7mcg/dL (normal 5-14mcg/dL).
The patient was started on supplementary growth hormone injections and hydrocortisone replacement with improvement in her dextrose levels. She was able to be weaned off of intravenous fluids and was able to maintain her dextrose levels with just oral feeding. The patient was discharged home on genotropin; hydrocortisone and synthroid with close follow up with endocrine, neurology and genetics. The final diagnosis was pan-hypopituitarism secondary to 18p deletion syndrome.
Discussion
18p deletion syndrome is a rare genetic syndrome first described in the literature in 1963 by de Grouchy and colleagues [1]. The syndrome is characterized by deletion of the whole or part of the short arm of the chromosome with worsening clinical symptoms the closer the deletion is to the centromere [2]. There have been many case reports since the early 1960s which report a constellation of symptoms associated with this syndrome which include cognitive impairments, small stature, minor facial dysmorphism, ptosis, strabismus, holoprosencephaly, speech and language difficulties, pituitary abnormalities and IgA deficiency. In particular the pituitary abnormalities can include a hypo plastic pituitary gland, an absent or ectopic posterior pituitary (Figure 1), complete absence of the pituitary gland or a hypo plastic pituitary stalk [1]. Our patient’s physical exam and diagnostic work up revealed dysmorphic facies and an ectopic posterior pituitary with chromosomal analysis confirming the diagnosis of 18p deletion syndrome.
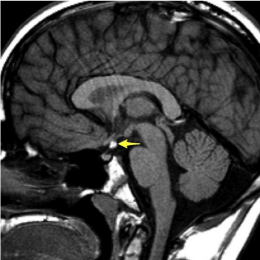
Figure 1: MRI of a child with ectopic posterior pituitary gland.
The patient’s presentation of refractory hypoglycemia was secondary to growth hormone deficiency due to her ectopic posterior pituitary gland. There are many reasons why an infant can present with hypoglycemia and understanding glucose metabolism both in a feeding and fasting state can help elucidate the diagnosis. After eating there is a rise in plasma glucose levels with a complementary rise in insulin levels. Insulin causes glucose uptake into the muscle and adipose tissue by increasing GLUT4 receptor expression. Insulin also promotes glycogen synthesis and inhibits glycogenolysis and hepatic gluconeogenesis [3]. When fasting, insulin expression is suppressed and glucagon stimulates glucose production from glycogen in the liver [1]. However glycogen stores can only last for 12-16 hours of fasting in a healthy child and when glucose levels become low the body releases counter-regulatory hormones to prevent hypoglycemia [4]. These hormones which include growth hormone and cortisol act to promote gluconeogenesis and activate lipolysis which increases utilization of fatty acids and reduces consumption of glucose [4]. If there is a hormone deficiency (such as in this patient) or lack of enzymes/substrates, the patient will have hypoglycemia and you may see the breakdown of triglycerides and the production of ketones in the blood and urine [3].
In the first three days of life, it is common to see low blood glucose levels in what is called ‘transitional neonatal hypoglycemia’. This is characterized by an initial fall in blood glucose levels from near maternal plasma levels to ~60mg/dL within the first two hours of life. Glucose levels then remain stable with little effect based on feeding style with a steady increase to ~80mg/dL by day 3-4 of life [5]. As stated earlier, this patient had low blood glucose levels in the first day of life requiring neonatal intensive care unit admission and it was likely believed at that time to be due to transitional neonatal hypoglycemia. However, when facing an older neonate or infant with persistent hypoglycemia it is imperative to acutely manage the symptoms and to further investigate an underlying diagnosis. Hypoglycemia can be especially harmful to the brain as the brain does not perform gluconeogenesis and has limited glycogen stores so requires a steady supply of glucose. Persistent hypoglycemia has been shown to be associated with acute phase occipitoposterior cortex edema and chronic phase atrophy. Long term neurological effects include visual problems, hearing loss, cognitive impairments and occipital lobe epilepsy [6].
In infants with persistent hypoglycemia, the workup should first include a detailed history and physical. It is important to know when the episode of hypoglycemia occurred and its relation to fasting. The physical exam can be suggestive of underlying syndromes associated with hypoglycemia [7]. Table 1 lists some of the more common physical exam findings which may indicate a genetic syndrome associated with hypoglycemia. Laboratory testing should be aimed at obtaining a “critical sample” while the patient is hypoglycemic and should include plasma glucose, bicarbonate, lactate, beta hydroxyl butyrate, free fatty acids and insulin.7While it is important to investigate for underlying metabolic disorders, it is important to not miss other medical emergencies including sepsis, meninogoencephalitis and toxic ingestion. Sepsis has been shown to cause hypoglycemia due to both cytokine induced glucose utilization and cytokine induced inhibition of gluconeogenesis. Particular ingestions which can lead to hypoglycemia include oral hypoglycemic agents such as sulfonylureas, cardiac medication such as beta-blockers, ethanol and salicylates. Table 2 summarizes the broad differential diagnosis for hypoglycemia that needs to be considered in an infant who presents with persistent hypoglycemia.
![]()
Clinical/genetic syndrome
Physical exam findings
Hypopituitarism
Micropenis
Cleft lip/palate
Short stature
Glycogenesis
Hepatomegaly
Adrenal insufficiency
Hyperpigmentation
Weight loss
Beckwith-Wiedemann
Hemihypertrophy
Omphalocele
Macroglossia
Table 1: Clinical/genetic syndromes associated with hypoglycemia.
![]()
Metabolic
Glycogen storage diseases
Fatty acid oxidation disorders
Hereditary fructose intolerance
Organic acidemias
Endocrine
Hyperinsulinemic hypoglycemia
Congenital adrenal hyperplasia
Autoimmune adrenal insufficiency
Hypopituitarism
Growth hormone deficiency
Infectious
Sepsis
Gastroenteritis
Malaria
Toxicology/Ingestion
Sulfonylureas
Beta blockers
Exogenous insulin
Ethanol
Table 2: Causes of hypoglycemia in an infant.
Once a hypoglycemic disorder has been identified, it is recommended that the plasma glucose concentration is maintained greater than 70mg/dl. Episodes of severe hypoglycemia should be treated with intravenous dextrose or use of glucagon in cases of hyperinsulinism [6,8]. In patients with hyperinsulinism, the aim of management is to avoid recurrent and occasionally unrecognized episodes of hypoglycemia in order to prevent the adverse neurological outcomes and sequelae. Long term therapy is based on the specific disorder that is recognized. For hyperinsulinsim, while focus on acute management is necessary, long term therapy can include surgery for those patients with refractory hypoglycemia in which the focal area of insulin overproduction in the pancreas is removed. For patients with growth hormone deficiency like the patient described in this case report, growth hormone supplementation allows for appropriate glucose counter regulation during episodes of hypoglycemia. For disorders which are fasting induced such as hereditary fructose intolerance, the management can be as simple as frequent feeding and high-carbohydrate diet.
Conclusion
There are many diagnoses to consider in an infant with persistent hypoglycemia. It is important to elicit a detailed history and physical exam and to understand the relationship of the hypoglycemia in relation to fasting. While acute management of the hypoglycemia needs to be prompt and may require admission to the intensive care unit, elucidating the underlying etiology will dictate long term management to avoid the neurologic sequelae of chronic hypoglycemia.
References
- Hasi-Zogaj M, et al. A review of 18p deletions. Am J Med Gen. 2015; 169: 251-264.
- Willoughby B, et al. Neuropsychological function in a child with 18p deletion syndrome: a case report. Cogn Behav Neurol. 2014; 27: 160-165.
- Ghosh A, et al. Recognition, assessment and management of hypoglycemia in childhood. Arch Dis Child. 2016; 101: 575-580.
- Kim S. Endocrine and metabolic emergencies in children: hypocalcaemia, hypoglycemia, adrenal insufficiency and metabolic acidosis including diabetic ketoacidosis. Ann Pediatr Endocrinol Metab. 2015; 20: 179-186.
- Stanley C, et al. Re-evaluating “transitional neonatal hypoglycemia”: mechanism and implications for management. J Pediatr. 2015; 166: 1520- 1526.
- Su J, et al. Research advances in neonatal hypoglycemic brain injury. Trans Pediatr. 2012; 1: 108-115.
- Thornton P, et al. Recommendations from the pediatric endocrine society for evaluation and management of persistent hypoglycemia in neonates, infants and children. J Pediatr. 2015; 167: 238-245.
- Gaillard F 2006. Ectopic posterior pituitary gland, MRI image, viewed June. 2016.