
Abstract
Trastuzumab, a humanized monoclonal antibody directed against human epidermal growth factor receptor 2 (HER2), remains the standard of care as part of adjuvant therapy for patients diagnosed with HER2-positive breast cancers. Despite high therapeutic efficacy, trastuzumab-based regimens can cause serious cardiotoxic side effects. Effective mitigation of cardiotoxic risk relies on thorough understanding of molecular mechanisms of cardiotoxicity induced by trastuzumab. Among the probable mechanisms responsible for trastuzumabmediated cardiotoxicity, generation of free radicals causing oxidative stress has garnered notable attention in recent years. More recently, role of autophagy in trastuzumab-induced cardiomyopathy was explored. Trastuzumab-mediated HER2 signaling dysregulation activated Erk/mTOR signaling cascade resulting in autophagy inhibition. Consequently, autophagy impairment leads to massive accumulation of damaged mitochondria and free radicals causing oxidative stress and toxicity in cardiomyocytes. This review will discuss recent advances in understanding the mechanism of oxidative stress and highlight the role of autophagy in trastuzumab-mediated cardiac dysfunctions.
Keywords: Trastuzumab; HER2; Autophagy; Reactive oxygen species (ROS); Cardiomyocytes; Doxorubicin; Cardiotoxicity
Introduction
Approximately 25-30% of breast cancers and 7-34% gastric cancers over express the Human Epidermal Growth Factor Receptor 2 (HER2) which is associated with poor prognosis of diseases [1-3]. Trastuzumab is an FDA-approved anti-HER2 monoclonal antibody, which is indicated for the treatment of HER2-overexpressing breast cancers and metastatic gastric or gastroesophageal junction adenocarcinoma [2, 4]. Trastuzumab selectively binds to extracellular domain IV of HER2 and inhibits HER-2-mediated mitogenic and pro-survival signaling pathways and proliferation of tumor cells [3]. In spite of being clinically highly efficacious, trastuzumab treatment is associated with severe cardiotoxicity in cancer patients. Trastuzumab-induced cardiac dysfunctions were first noticed in a pivotal phase III clinical trial, which reported that 27% of patients receiving anthracyclines, cyclophosphamide and trastuzumab exhibited cardiovascular events, including asymptomatic decline in Left Ventricular (LV) systolic function, suggesting that addition of trastuzumab increased chemotherapy-associated symptomatic and asymptomatic cardiac dysfunction [5].
Trastuzumab-related cardiotoxicity has been proposed as a type II treatment-related cardiovascular disorders, which are largely reversible in nature and do not cause primary cardiomyocyte destruction [6,7]. However, there are clinical reports that have indicated that cardiotoxicity induced by trastuzumab may not be reversible in some of trastuzumab-treated patients, hence raising the skepticism over reversibility concept of trastuzumab-induced cardiotoxicity [8]. Mechanisms behind trastuzumab-mediated cardiotoxicity are still not well understood. In this mini-review, we discuss recent advances in understanding of molecular mechanism of trastuzumab-induced cardiotoxicity and focus on autophagy dysregulation as potential mechanism for oxidative stress caused by trastuzumab.
Oxidative Stress and Trastuzumab-Induced Cardiotoxicity
Despite the considerable efforts to divulge the molecular aspects of trastuzumab-induced cardiotoxicity, mechanisms still remain elusive. Constitutively active cardiomyocytes require high energy in the form of ATP, which is produced from mitochondria with the generation of Reactive Oxygen Species (ROS) in cardiomyocytes. Under normal physiological conditions, endogenous antioxidants are capable of clearing out majority of ROS species, thus maintaining the cardiomyocytes in stress-relieved conditions. HER2 signaling, which is pivotal for cardiomyocyte survival and global cardiac functions, activates prosurvival Mitogen-Activated Protein Kinase (MAPK/Erk) signaling and Phosphoinositide 3 Kinase (PI3K/AKT) to maintain ATP levels and keep ROS levels at low concentration in cardiomyocytes [9]. Growing evidences from pre-clinical models have established that treatment with trastuzumab causes oxidative and nitrative stress in myocardium by interfering with HER2 signaling and inhibiting its pro-survival effects. In rat cardiomyocyte model, blockade of HER2 signaling pathway using anti-HER2 antibody induced cardiomyocyte death via mitochondrial and ROS-dependent pathways [10].Using C57/BL6 mice model of trastuzumab toxicity, we have previously demonstrated that 3- Nitrotyrosine (NT) and 4- Hydroxynonenal (4-HNE), which are byproducts of oxidative and nitrative damage in cardiomyocytes, were significantly elevated in the heart tissues of trastuzumab-treated animals [11]. Additionally, we showed that trastuzumab treatment impacts on the expression of myocardial genes essential for cardiac and mitochondrial function, and causes ultrastructural damages of myocardium, thus compromises the cardiomyocyte viability and increase oxidative stress [11]. Based on these mechanistic insights, it is evident that when HER2 signaling is blocked by trastuzumab, cardiomyocytes are unable to activate protective survival pathways to cope with excess radical species production, leading to disruption of cellular integrity which causes cardiac dysfunctions [12].
Neuregulin (NRG1/ErbB) signaling is another potential pathway that interferes with oxidative stress conditions of myocardia by activating HER4, which then forms heterodimer with HER2 to initiate cardiomyocyte survival pathways. NRG1-mediated tyrosine kinase activation and G-Protein coupled receptor signaling primarily activates three major cardiomyocyte survival cascades: MAPK/Erk, PI3K/AKT, and Focal Adhesion Kinases (FAKs), all of which are required for cardiomyocyte survival by inhibiting ROS production [13,14]. When trastuzumab binds to HER2, it compromises its ability to form homo or heterodimer with other HER family receptors and subsequent activation of survival signaling, thereby adversely affecting the cardio-protective properties of NRG1 [15,16]. Renin angiotensin system is NRG1-dependentsignaling pathway involved with pathogenesis of drug-induced cardiotoxicity and oxidative damage. Angiotensin (Ang I/II) signaling is upregulated in the heart during stress conditions and exhibits its deleterious side effects via two mechanisms. Firstly, Ang I inhibits cardio-protective properties of NRG1 by suppressing the ability of NRG1 to bind with HER receptor, leading to inhibition of the essential MAPK/Erk cell survival pathways, which further accelerates ROS production. Secondly, Ang II enhances the levels of Nicotinamide Adenine Dinucleotide Phosphate Oxidase (NADPH) through binding to Ang II type 1 receptor (AT1) receptors [17]. The Ang II and AT1 interaction contributes towards activation of specific apoptosis pathways and causes oxidative damage to cardiac structure. In spite of recent advances in understanding of trastuzumab-mediated oxidative stress mechanisms, a direct correlation between trastuzumab-induced cardiomyopathy and impaired HER2 signaling leading to increased ROS production have not been established [7]. In next sections, we will discuss significance of autophagy process in cardiac development and elaborate the novel role of autophagy as a possible link between dysregulated HER2 signaling and trastuzumab-induced oxidative stress in cardiac models.
Autophagy Signaling in Cardiovascular Diseases
Autophagy plays an essential and highly conserved role in normal heart development in vertebrate. Knockdown of essential autophagy regulatory genes results in abnormal cardiac development including defects in morphogenesis, abnormal heart structure, and reduced organismal survival in zebrafish model [18]. Mammalian autophagy is primarily governed by a serine/threonine kinase mTOR, which negatively regulate autophagy during nutrient rich conditions. Under nutrient rich conditions, mTORC1 suppress autophagy by phosphorylating the autophagy-initiating UNC-5 like autophagy activating kinase (ULK1) at Ser758, thereby preventing its activation by AMPK, a key activator of autophagy [19]. Studies have demonstrated that upstream and downstream signaling molecules of mTOR can regulate autophagy initiation and progression. Ribosomal protein S6, a direct and most studied downstream substrate of mTOR signaling, can suppress autophagic proteolysis in the presence of amino acid, suggesting that autophagy inhibition and S6 activation are under the control of mTOR signaling [20]. Upstream regulator of mTOR signaling, MAPK/Erk can also regulate expression of autophagy genes and a recent study showed that EGF treatment facilitates the interaction of Erk cascade components with autophagy related proteins in the cytosol and nucleus [21].
A basal level of autophagy is essential for cellular homeostasis in cardiomyocytes, which can play protective role by recycling damaged proteins, glycogens and fatty acid, thus supplying the energy to myofiber in response to nutrition deprivation, growth factor depletion and hypoxia. Autophagy functions as an important pathophysiological response in heart patients suffering from various cardiomyopathies including coronary artery disease, hypertension, aortic valvular disease, and congestive heart failure. In a study, Nakai et al. reported that deletion of temporary controlled cardiac-specific Atg5 (autophagy related protein-5) resulted in left ventricular dilatation, severe contractile dysfunction, disorganized sarcomere structure, and accumulation of damaged mitochondria in adult mice model [22]. This study concluded that constitutive autophagy is protective mechanism for maintaining structural and functional integrity of cardiomyocytes, and inhibition of autophagy could potentially lead to progression of cardiac hypertrophy and cardiomyopathy. Similarly, very early studies also reported the importance of autophagy inhibition in progression of cardiomyocyte hypertrophy [23]. Mice deficient in crucial autophagy regulator LAMP-2 (lysosome associated membrane protein-2) showed excessive accumulation of autophagic vacuoles and abnormal cardiomyocyte ultrastructure, further highlighting the critical involvement of autophagy-lysosomal pathway in cardiomyopathy and heart disorders [24]. In desminrelated cardiomyopathy model, autophagy inhibition by inactivating Beclin-1 gene expression dramatically enhanced accumulation of intracellular aggregates, accelerated ventricular dysfunction and heart failure, suggesting that autophagy upregulation is adaptive response in proteotoxic form of cardiac disease [25]. Usage of autophagy inhibitor 3-methyladenine in THP1 macrophages resulted in accumulation of damaged mitochondria and increased ROS production, emphasizing the significance of autophagy regulation in pathogenesis of oxidative stress conditions [26]. These studies provide ample evidence of critical role of autophagy in maintaining global cardiac functions.
Autophagy Dysregulation in Trastuzumab- Induced Cardiovascular Toxicities
A number of studies have endorsed the integral role of HER2 signaling in cardiac development and cardiomyocytes survival [9,27]. Cardiac-specific HER2 knock-out mice develop enlarged cardiac chambers, reduced muscle contractility and thinner cardiac walls, resulting in dilated cardiomyopathy. A recent study by Mohan et al. revealed a novel mechanism by which trastuzumab binding to domain IV of HER2 in cardiomyocytes impairs HER2 signaling, leading to suppression of autophagy and enhanced oxidative stress in cardiomyocytes [28]. This study, for the first time, unraveled a mechanism by which trastuzumab-induced dysregulated HER2 signaling activates mTOR/Ulk1 kinase activity and mediates the inhibition of autophagy in cardiomyocytes. Particularly, trastuzumab induces phosphorylation of HER1 at 845 and HER2 at 1248 sites in human primary cardiomyocytes, which subsequently activates Erk/mTOR signaling pathway in cardiac cells [28]. Trastuzumabmediated Erk/mTOR activation induced time-dependent decrease in autophagy by down regulating LC3 I/II expression and increasing p62 levels. Trastuzumab also dismantled autophagosome-assembly machinery by inhibiting Atg 5-12, Atg 7, Atg 14, and Beclin 1, the key molecules involved with autophagosome complex proteins. Since autophagy is primarily responsible for recycling of damaged organelles and unwanted protein aggregates in heart, the autophagy suppression induced by trastuzumab resulted in accumulation of damaged cellular materials. Mohan et al. further demonstrated that trastuzumab treatment in cardiomyocytes significantly enhanced generation of chemically reactive species, peroxides and free radicals, thus rendering the myocardium to more susceptible for oxidative damage [28]. The mechanistic association between trastuzumab-induced HER signaling pathways and subsequent onset of cardiotoxicity has been presented in Figure 1. Many studies have shown that ROS function as upstream regulator of autophagy under nutrient deprivation conditions; however, autophagy blockade can result in accumulation of damaged mitochondria and increased generation of mitochondrial ROS, suggesting that autophagy activity can be upregulated to remove source of toxic ROS [26]. Autophagy in cardiovascular biology and the signaling pathways regulating mechanisms of ROS/RNS generation, role of autophagy in response to oxidative stress and alternative mechanisms by which autophagy regulates mitochondrial functions have been thoroughly reviewed elsewhere [29,30]. Future studies are warranted to fully understand the complex interplay between ROS and autophagy signaling which will assist in development of effective therapeutic approach against trastuzumab and chemotherapy induced cardiotoxicities.
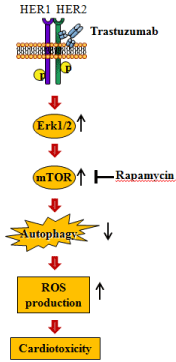
Figure 1: Schematic representation illustrates a proposed model depicting
the relationships between trastuzumab-induced HER1/HER2 phosphorylation
and Erk/mTOR upregulation, and autophagy inhibition leading to enhanced
ROS production and cardiotoxicity. Rapamycin may potentially reduce
trastuzumab-induced cardiotoxicity by inhibiting mTOR activity and, therefore,
reducing ROS production.
It has been widely debated whether trastuzumab-mediated HER2 signaling in breast cancer models is distinct from cardiac models. In our previous investigation, Dokmanovic et al., demonstrated that binding of trastuzumab to HER2 activates HER2 kinase activity which partially contributes to enhanced HER2 phosphorylation at 1248 in trastuzumab-sensitive SKBR3 and BT474 breast cancer cells [31]. Nevertheless, downstream of HER2 signaling appears to be different in HER2-positive breast cancers and cardiomyocyte system because trastuzumab, which significantly suppress Akt in SKBR3 and BT474 cells, does not affect Akt signaling in human cardiomyocytes. Interestingly, rapamycin which is a potent mTOR inhibitor and prominent autophagy inducer, can rescue the cardiomyocytes from trastuzumab-induced oxidative stress, highlighting the clinical benefit of mTOR inhibitors in trastuzumab-based regimens [28]. Inhibition of mTOR signaling by rapamycin has been shown to prevent cardiac hypertrophy and improve cardiac function in pressure-overload rats [32].
Several clinical trials have confirmed the synergistic potentiation of cardiotoxic effects of trastuzumab when it is administered following doxorubicin therapy as concurrent or sequential regimen [5,33]. Doxorubicin has inherent ability to increase oxidative stress in cardiomyocytes [34]. Mechanistic study discovered that doxorubicin can significantly increase cellular ROS level through Nox2 NADPH oxidase-mediated biochemical reactions and contribute towards the pathogenesis of cardiac contractile dysfunctions and remodeling [34]. When trastuzumab is used in conjunction with doxorubicin therapy, it can block the key cell proliferation pathways and enhance ROS production in cardiomyocytes [12]. Although doxorubicin and trastuzumab can induce similar toxic effect in cardiomyocytes, their effects are classified into different categories of cardiac damage: doxorubicin is a type I agent, featuring cumulative and dose-related myocyte permanent injury or death; while trastuzumab is a type II agent, featuring no cumulative, no dose-related and reversible myocyte dysfunction [7]. It is generally believed that the concomitant administration of these two drugs should be avoided due to potentially additive or synergistic increase in the risk of cardiomyopathy. Recently, role of autophagy in doxorubicin-induced cardiotoxicity has been explored; however, conflicting results have been reported regarding the effect of doxorubicin on autophagy and its role in cardiotoxicity [35]. Doxorubicin treatment enhanced autophagy in vitro and in vivo in rat cardiac models, but suppressed autophagy in mouse cardiac models, suggesting that effect of doxorubicin on autophagy may be species specific [35]. Regardless of doxorubicin’s impact on cardiac autophagy, it is unclear how trastuzumab would affect autophagic pathways in cardiac models when it is dispensed as concurrent or sequential therapy followed by doxorubicin.
Conclusion
Cardiotoxicity of trastuzumab is a major risk for HER2-positive breast cancer patients undergoing therapy and surviving cancer attack. It is also a critical factor limiting the use of this important drug on HER2-positive breast cancer patients. Mechanisms of cardiotoxicity induced by trastuzumab are evolving continuously. Autophagy and ROS are closely related mechanisms critical for maintaining normal and active cardiomyocyte function; therefore further research is warranted to investigate the clinical benefit of autophagy modulators in trastuzumab-based therapies. Combining mTOR inhibitors with trastuzumab for cancer treatment may not only show better therapeutic response against tumor proliferation but also protect heart from trastuzumab-induced cardiac risks. Effective mitigation of cardiotoxic risk with trastuzumab relies on thorough understanding of its binding effect on HER2 signaling, the downstream of HER2 signaling in human cardiomyocytes, and related interference to the immune system. Exploring new chemical and biological entities to manipulate these pathways are critical for breast cancer patients.
Grant Support
This work was supported by FDA Office of Women’s Health Research Science Program award to Wen Jin Wu.
Acknowledgement
This project was supported in part by an appointment to the ORISE Research Participation Program at the Center for Drug Evaluation and Research (CDER), U.S. Food and Drug Administration, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and FDA. Dr. Nishant Mohan is an ORISE research fellow supported by Food and Drug Administration Office of Women’s Health. The authors thank Drs. Haoheng Yan and Harsh V. Jain for critical internal review of the manuscript.
Disclaimer
This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
References
- Yu D, Hung MC. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene. 2000; 19: 6115-6121.
- Ruschoff J, Hanna W, Bilous M, Hofmann M, Osamura RY, Penault-Llorca F, et al. HER2 testing in gastric cancer: a practical approach. Mod Pathol. 2012; 25: 637-650.
- Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med. 2007; 357: 39-51.
- Loibl S, Gianni L. HER2-positive breast cancer. Lancet. 2016.
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001; 344: 783-792.
- Ewer MS, Vooletich MT, Durand JB, Woods ML, Davis JR, Valero V, et al. Reversibility of trastuzumab-related cardiotoxicity: new insights based on clinical course and response to medical treatment. J Clin Oncol. 2005; 23: 7820-7826.
- Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol. 2015; 12: 547-558.
- Telli ML, Hunt SA, Carlson RW, Guardino AE. Trastuzumab-related cardiotoxicity: calling into question the concept of reversibility. J Clin Oncol. 2007; 25: 3525-3533.
- Negro A, Brar BK, Lee KF. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog Horm Res. 2004; 59: 1-12.
- Gordon LI, Burke MA, Singh AT, Prachand S, Lieberman ED, Sun L, et al. Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial and reactive oxygen species-dependent pathways. J Biol Chem. 2009; 284: 2080-2087.
- ElZarrad MK, Mukhopadhyay P, Mohan N, Hao E, Dokmanovic M, Hirsch DS, et al. Trastuzumab alters the expression of genes essential for cardiac function and induces ultrastructural changes of cardiomyocytes in mice. PLoS One. 2013; 8: 79543.
- Zeglinski M, Ludke A, Jassal DS, Singal PK. Trastuzumab-induced cardiac dysfunction: A ‘dual-hit’. Exp Clin Cardiol. 2011; 16: 70-74.
- Pentassuglia L, Sawyer DB. The role of Neuregulin-1beta/ErbB signaling in the heart. Exp Cell Res. 2009; 315: 627-637.
- Jiang Z, Zhou M. Neuregulin signaling and heart failure. Curr Heart Fail Rep. 2010; 7: 42-47.
- Pentassuglia L, Timolati F, Seifriz F, Abudukadier K, Suter TM, Zuppinger C. Inhibition of ErbB2/neuregulin signaling augments paclitaxel-induced cardiotoxicity in adult ventricular myocytes. Exp Cell Res. 2007; 313: 1588- 1601.
- Rochette L, Guenancia C, Gudjoncik A, Hachet O, Zeller M, Cottin Y, et al. Anthracyclines/trastuzumab: new aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol Sci. 2015; 36: 326-348.
- Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003; 35: 851-859.
- Lee E, Koo Y, Ng A, Wei Y, Luby-Phelps K, Juraszek A, et al. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 2014; 10: 572-587.
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13: 132-141.
- Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995; 270: 2320-2326.
- Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, Singh R. Autophagy proteins regulate ERK phosphorylation. Nat Commun. 2013; 4: 2799.
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007; 13: 619-624.
- Dammrich J, Pfeifer U. Cardiac hypertrophy in rats after supravalvular aortic constriction. II. Inhibition of cellular autophagy in hypertrophying cardiomyocytes. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983; 43: 287- 307.
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2- deficient mice. Nature. 2000; 406: 902-906.
- Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci USA. 2008; 105: 9745-9750.
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011; 469: 221-225.
- Iwamoto R, Mekada E. ErbB and HB-EGF signaling in heart development and function. Cell Struct Funct. 2006; 31: 1-14.
- Mohan N, Shen Y, Endo Y, ElZarrad MK, Wu WJ. Trastuzumab, but Not Pertuzumab, Dysregulates HER2 Signaling to Mediate Inhibition of Autophagy and Increase in Reactive Oxygen Species Production in Human Cardiomyocytes. Mol Cancer Ther. 2016; 15: 1321-1331.
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009; 43: 67-93.
- Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest. 2015; 125: 55-64.
- Dokmanovic M, Wu Y, Shen Y, Chen J, Hirsch DS, Wu WJ. Trastuzumabinduced recruitment of Csk-homologous kinase (CHK) to ErbB2 receptor is associated with ErbB2-Y1248 phosphorylation and ErbB2 degradation to mediate cell growth inhibition. Cancer Biol Ther. 2014; 15: 1029-1041.
- McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004; 109: 3050-3055.
- Thavendiranathan P, Abdel-Qadir H, Fischer HD, Camacho X, Amir E, Austin PC, et al. Breast Cancer Therapy-Related Cardiac Dysfunction in Adult Women Treated in Routine Clinical Practice: A Population-Based Cohort Study. J Clin Oncol. 2016; 34: 2239-2246.
- Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, et al. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010; 70: 9287-9297.
- Dirks-Naylor AJ. The role of autophagy in doxorubicin–induced cardiotoxicity. Life Sci. 2013; 93: 913–916.