
Abstract
Cardiovascular disease remains the leading cause of death for both men and women in the United States despite the recent advances in drug development, changes in lifestyle and screening protocols. A key target in the treatment of cardiovascular disease is the Renin-Angiotensin System (RAS) and pharmacologic approaches have traditionally focused on the Ang II-AT1 receptor axis of the RAS to prevent the generation of Ang II with Angiotensin Converting Enzyme Inhibitors (ACEI) or to block the binding of Ang II to the AT1 Receptor (AT1R) with selective antagonists (ARBs). The RAS, however, exhibits an alternative ACE2-Ang-(1-7)-AT7/Mas receptor pathway in the circulation and in both central and peripheral tissues that generally opposes the Ang II-AT1R axis and whose actions include stimulation of nitric oxide and attenuation of oxidative stress. The brief review examines the various approaches that would target the alternative Ang-(1-7) axis of the RAS as a therapeutic treatment of cardiovascular disease and other pathologies.
Keywords: Angiotensin; Betamethasone; Diabetic mice
Introduction
Cardiovascular disease remains the leading cause of death for both men and women in the United States despite the recent advances in drug development, changes in lifestyle and screening protocols. A key target in the treatment of cardiovascular disease and hypertension is the Renin-Angiotensin System (RAS), a circulating and tissue system involved in the regulation of blood pressure, fluid balance and cellular injury. Pharmacologic approaches have traditionally focused on the Ang II-AT1 receptor axis of the RAS to prevent the generation of Ang II with Angiotensin Converting Enzyme Inhibitors (ACEI) or to block the binding of Ang II to the AT1 Receptor (AT1R) with selective antagonists (ARBs). Beginning with our demonstration of endogenous levels of the heptapeptide peptide Ang-(1-7) in the circulation, brain and other relevant cardiovascular tissues, alternative pathways within the RAS are present that generate bioactive peptides other than Ang II [1]. The concept of an alternative RAS with Ang-(1-7) as the principal bioactive component was bolstered by identification of the Mas receptor that mediates the actions of Ang-(1-7) by Santos, Bader, Thomas and colleagues, as well as the ACE homolog ACE2 capable of directly converting Ang II to Ang-(1-7) by the Acton, Turner, and Penninger labs [2-5]. Ang-(1-7) exhibits distinct actions to functionally oppose the deleterious effects of an activated ACE-Ang II-AT1R axis [6]. Experimental evidence to date suggests that Ang- (1-7) exhibits a wide range of cardioprotective effects that involve the stimulation of the Nitric Oxide Synthase (NOS) and release of NO, the attenuation of oxidative stress and a reduction in inflammatory signaling [7]. The current review examines the various approaches that would target the alternative an-(1-7) axis in the treatment of cardiovascular disease and other pathologies including cancer, diabetes and tissue fibrosis [7].
Angiotensin-(1-7) Formation
Circulating Ang-(1-7) is derived from the inactive precursor peptide Ang I by the Metallo Endo Peptidase (MEP) neprilysin (EC3.4.24.11) [8]. Although there is little to no circulating neprilysin, the endopeptidases is anchored to the cell membrane with the active site orientated to the extracellular space that allows for the processing of circulating Ang I to Ang-(1-7). Apart from the circulation, Ang-(1- 7) expression is evident in many tissues and may reflect the processing of Ang I by an intracellular MEP- thimet oligopeptidase (EC3.4.24.15) [8]. Both endopeptidases efficiently hydrolyze the Pro7-Phe8 bond of Ang I to directly generate Ang-(1-7); however, neprilysin also cleaves of the Tyr4-Ile5 of Ang I to form Ang-(1-4) [8]. Nagata and colleagues have identified an extended form of Ang I termed Ang-(1-12) in plasma and various tissues in rat that may reflect renin-independent processing of the RAS precursor angiotensinogen [9]. We originally found that Ang-(1-12) is also a substrate for neprilysin to generate Ang-(1-7); however, Ang-(1-12) is converted to Ang II by successive hydrolysis of Tyr10-Tyr11 and Phe8-His9 bonds via ACE [10]. This is likely the predominant processing pathway for Ang-(1-12) in the circulation given the high levels of ACE and that the increase in blood pressure to Ang-(1-12) infusion was abolished by an ACE inhibitor [9]. Endogenous levels of Ang-(1-7) may also originate from processing of Ang II by the mono-carboxypeptidases angiotensin converting enzyme 2 (ACE2, EC3.4.17.23). prolyl oligopeptidase (EC3.4.21.26), and prolyl carboxypeptidases (EC3.4.16.2) [8]. These pathways initially require the traditional processing of Ang I by ACE to generate Ang II as the substrate for the conversion to Ang-(1- 7). Santos and colleagues also identified an endogenous isoform of Ang-(1-7) in human plasma that arises from decarboxylation of the aspartic acid residue to alanine to form [Ala1]-Ang-(1-7) [11]. The peptide appears to share similar properties to Ang-(1-7); however, [Ala1]-Ang-(1-7) may preferentially bind to the Mas-Related G protein-coupled receptor D (MRG-D) rather than the MasR. Moreover, both the AT7/MasR antagonist [D-Pro7]-Ang-(1-7) and the AT2R antagonist PD123319, but not [D-Ala7]-Ang-(1-7) block the vasorelaxant effects of the peptide [11]. It is presently unknown whether Ang-(1-7) itself is decarboxylated or that existing alanine isoforms of Ang II and Ang I are processed to [Ala1]-Ang-(1-7).
ACE2
ACE2 is an extracellularly-oriented monocarboxypeptidase that converts Ang II to Ang-(1-7) and the enzyme is widely distributed in various tissues [8]. We previously showed that ACE2 was the primary enzymatic pathway in the mouse heart for the formation of Ang-(1-7) from Ang II consistent with studies in the human heart [12,13]. The peptidase is now considered a key target of the RAS as the altered expression of the enzyme may influence the balance of Ang II and Ang-(1-7) [8]. Raizada and colleagues initially recognized the importance of this regulatory relationship and identified two allosteric activators of ACE2 (xanthenone and diminazene aceturate) that increase peptidase activity about 2-fold [14]. Administration of these small molecule activators significantly reduced blood pressure and attenuated tissue fibrosis; these effects were reversed by [D-Ala7]- Ang-(1-7) antagonist suggesting a primary action of Ang-(1-7) rather than Ang II [15-17]. However, the cardioprotective effects of ACE2 activators have not been consistently replicated by other investigators which may in part reflect different models of cardiovascular disease or tissue injury [18-20]. Moreover, it is now apparent that these activators may exhibit other properties that are distinct from an allosteric effect to augment the catalytic rate of ACE2 [18-20].
An alternative approach to pharmacologically modifying ACE2 activity is the direct administration of a soluble form of recombinant ACE2 (rACE2) that retains the specificity and catalytic activity of the native enzyme [21,22]. The premise of ACE2 supplementation is that sufficiently high levels of ACE2 are administered to reduce Ang II/Ang-(1-7) thereby attenuating the actions of Ang II-AT1R axis while amplifying the Ang-(1-7)-AT7/MasR effects (Figure 1). Oudit and colleagues find that chronic administration of soluble rACE2 attenuated various indices of cardiac and renal injury, inflammation and fibrosis in both type 1 and type 2 diabetic mice [23,24]. Surprisingly, the administration of rACE2 reduced tissue levels of Ang II in the heart and kidney and increased the tissue contents of Ang-(1-7) [23]. It is unclear how increased circulating rACE2 augments Ang-(1-7) tissue levels as the intracellular mechanism for Ang II or Ang-(1-7) generation is not known. One possibility is that administered rACE2 increases circulating levels of Ang-(1- 7) and the peptide is subsequently internalized by the Mas receptor into a stable or protected intracellular compartment. In turn, rACE2 treatment may reduce circulating levels of Ang II that would lead to less internalization of the Ang II-AT1R complex and reduced tissue content. Alternatively, rACE2 treatment may alter that intracellular signaling milieu to attenuate oxidative stress or inflammation that impacts the local generation of Ang II and Ang-(1-7). Scholey and colleagues also report that rACE given subcutaneously by an osmotic pump attenuated several indices of renal damage in the transgenic Col4A3-/- mouse, a model of Alport syndrome, as well as tended to lower blood pressure [25]. The renal protective effects of soluble rACE2 treatment were associated with a marked reduction in the ratio of Ang II to Ang-(1-7) in the kidney have (from 69 to 15) while the wild type Ang II/Ang-(1-7) value was 4 (peptide values were corrected to fmol/mg protein for ratio determination) [25]. However, Wysocki et al observed that neither the administration of rACE2 nor the chronic overexpression of the soluble peptidase by mini circle DNA conveyed any protective effects against diabetic nephropathy in diabetic mice [26]. In this study, the plasma angiotensin peptides were quantified by UHPLC-mass spectroscopy with sufficient sensitivity to detect peptides in the low pM range and resolve related N-terminal metabolites of Ang I, Ang II and Ang-(1-7) that include Ang-(2-10), Ang-(2-8), Ang-(3-8), Ang-(2-7) and Ang-(3- 7), respectively; these metabolites are not distinguished by direct RIAs or ELISAs unless coupled to HPLC/UHPLC separation prior to assay. This analysis revealed that chronic ACE2 treatment reduced the total plasma ratio of Ang II to Ang-(1-7) approximately 4-fold in the diabetic mice; however, the effect of ACE2 on renal peptide content was not addressed. Moreover, the fact that the N-terminal metabolites Ang-(2-10), Ang-(2-8) or Ang III and Ang-(2-7) were the major components in plasma contrasts with the accepted profile of angiotensins in both the circulation and tissues [8].
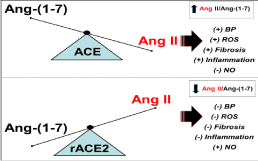
Figure 1: Scheme depicting the influence of exogenous ACE2 on the ratio of Ang II to Ang-(1-7). Upper panel: Predominance of ACE activity increases the ratio of
Ang II to Ang-(1-7) that may contribute to increased blood pressure (+BP), oxidative stress (ROS), fibrosis and inflammation, but reduced levels of nitric oxide (-NO).
Lower panel: Treatment with ACE2 or overexpression of the peptidase reduces the Ang II to Ang-(1-7) ratio that may lower BP, ROS, fibrosis and inflammation,
but increase levels of NO.
Although the disparate findings of rACE2 treatment to ameliorate renal injury are difficult to reconcile, the concept of ACE2 administration to regulate the endogenous RAS may present several disadvantages. One concern is that a reduction in Ang II and the subsequent lowering of blood pressure by exogenous ACE2 should markedly stimulate the conventional RAS as the negative feedback mechanisms on renin are removed or disinhibited. Elevated renin will generate Ang I, and given the very high ACE content in the vasculature, Ang I is immediately converted to Ang II to increase the peptide. The second issue is since ACE is the major pathway for the metabolism of Ang-(1-7), it appears unlikely that exogenous ACE2 would result in a sustained increase in Ang-(1-7) unless ACE activity is sufficiently blocked or down regulated. ACE readily hydrolyzes the Ile5-His6 bond of Ang-(1-7) to generate the dipeptide His-Pro and Ang-(1-5) which explains the increased circulating levels of Ang-(1- 7) with ACE inhibitors [8]. We suggest that a more effective approach would be the administration of ACE2 combined with an ACE inhibitor to enhance the metabolism of residual levels of Ang II and prevent the degradation of the Ang-(1-7). Importantly, this approach would not impact the endogenous pathway for the conversion of Ang I [or Ang-(1-12)] to Ang-(1-7) by neprilysin or thimet oligopeptidase as the Ang II substrate is depleted.
Angiotensin-(1-7) Analogs
An alternative approach to increase endogenous levels of Ang- (1-7) by ACE2 is the direct administration of the peptide. However, the native peptide is rapidly metabolized by ACE in the circulation and cannot be administered orally. To circumvent the bioavailability issue, investigators at Aventis Pharmaceutical synthesized the first non-peptide agonist (AVE 0991) to the AT7/MasR that is orally active and is likely not a substrate of ACE [27]. AVE 0991 appears to share the same properties of Ang-(1-7) and the actions of the analog are blocked by the AT7/MasR antagonists [D-Ala7]-Ang-(1-7) and [D-Pro7]-Ang-(1-7) [28]. Santos and colleagues have developed a different approach in which Ang-(1-7) is encapsulated within a cyclodextrin complex that is orally available and conveys similar biological actions as Ang-(1-7), although it’s unclear whether the cyclodextrin form of Ang-(1-7) is more resistant to metabolism than the native peptide [29]. Haas and colleagues synthesized a peptide analog of Ang-(1-7) in which the peptide contained a thioether bridge between the substituted residues Ser4 and Cys6 [30]. This peptide analog is biologically active and is protected from ACE hydrolysis by the bridged residue at the 6th position [30,31]. We recently reported the effects of single substitutions along the Ang-(1- 7) peptide chain using novel non-natural d-Amino Acids Derivatives (ACCA) on peptide metabolism and inhibition of cell growth. ACCA substitutions to Iso5 or His6 of Ang-(1-7) essentially abolished the metabolism of the peptide analog by human ACE, as well as retained the anti-proliferative actions of the peptide in two tumor cell lines [32]. Interestingly, the ACCA substitution of Val3 did not prevent ACE metabolism, but did abolish N-terminal hydrolysis by dipeptidyl amino peptidase 3 (EC3.4.14.4; DAP 3). DAP 3 hydrolyses Ang-(1-7) to Ang-(5-7) in a 2-step reaction that initially generates the dipeptide Asp-Arg and Ang-(3-7), and then rapidly cleaves Ang- (3-7) to the dipeptide Val-Tyr and the tripeptide Ile-His-Pro [33]. We previously reported that DAP 3 activity was increased 3-fold in the Cerebrospinal Fluid (CSF) of adult sheep that were exposed to betamethasone in utero as a model of fetal programming to hasten pulmonary development of premature infants [7]. DAP 3 activities in the CSF increased to a greater extent than ACE in the betamethasone betamethasoneexposed sheep and both peptidases may contribute to the lower CSF levels of Ang-(1-7) and an altered baroreflex control of blood pressure [7]. DAP 3 activity was also evident in the intracellular and extracellular compartments of human HK-2 proximal tubule cells [34]. In these cells, treatment with the MEP inhibitor JMV-390 attenuated DAP 3 activity and increased the intracellular levels of Ang-(1-7) approximately 2-fold; however, a higher concentration of JMV-390 significantly reduced Ang-(1-7) in the HK-2 cells that likely reflects crossover of the inhibitor to block thimet oligopeptidase to reduce Ang-(1-7) generation [33].
Finally, we note that a non-peptide orally available agonist of the AT2R (C21) was recently developed and exhibits cardioprotective effects similar to the Ang-(1-7) pathway. Katovitch and colleagues find that C21 attenuated the extent of pulmonary fibrosis and pulmonary hypertension [35]. These protective effects of C21 were abolished by the AT2R antagonist PD123319; however, the C21 effects were also blocked by the [D-Ala7]-Ang-(1-7) antagonist [35]. It is presently unclear whether the C21 compound directly stimulates the AT7/MasR or that activation of the AT2R requires the interaction or transactivation of the Ang-(1-7) receptor; however, both the AT2R and AT7/MasR were required to transducer the effects of either C21 or Ang-(1-7) to stimulate expression of the chemokine receptor-1 in astrocytes [36]. We also find that the potent vasorelaxation of the renal artery by Ang-(1-7) (ED50 of 3nM) was blocked by [D-Ala7]- Ang-(1-7) and the AT2R antagonist PD123319, as well as the soluble guanylate cyclase inhibitor ODQ [37]. Thus, it is feasible that the AT2R agonist C21 may also stimulate the Ang-(1-7)-AT7/MasR axis and that C21 could be combined with conventional RAS therapies including ACEIs or ARBs.
Conclusion
In conclusion, activation of the Ang-(1-7)-AT7/MasR axis may be a potentially important therapeutic target in the treatment of cardiovascular disease. The continued development of selective non-peptide analogs of Ang-(1-7) that target both Mas and MRG-D receptors may be of particular benefit in terms of their oral availability, greater resistance to peptidase metabolism and their potential to accumulate within the cell. Indeed, we demonstrated the expression of intracellular AT7/Mas receptors on isolated nuclei from the ovine kidney that were linked to NO generation [38]. Moreover, the AT7/MasR binding density on renal nuclei and the Ang-(1-7)- evoked NO response was reduced in aged sheep and those exposed to Betamethasone in utero as compared to the younger non-exposed animals [7,38]. Conversely, intracellular levels of the AT1R that were associated with stimulation of oxidative stress on isolated nuclei were increased in the older animals and in the Betamethasone-exposed sheep [7,38]. These latter findings of nuclear AT1R on the ovine renal cortex support extensive evidence for an intracellular RAS within various tissues although the exact role of an activated intracellular RAS to influence cardiovascular disease and other pathologies is unknown and awaits more conclusive studies [38-40]. Nevertheless, peptidase resistant and cell permeable agonists of the Ang-(1-7) axis may provide additional cardioprotective effects to conventional approaches to block the RAS.
Funding
These studies were supported in part by grants from the National Institute of Health grants (HL-56973, HL-51952, HD084227, HD- 047584 and HD-017644 and the American Heart Association (AHA- 151521 and AHA-355741). An unrestricted grant from the Farley- Hudson Foundation (Jacksonville, NC), Groskert Heart Fund and the Wake Forest Venture Fund is also acknowledged.
References
- Chappell MC, Brosnihan KB, Diz DI, Ferrario CM. Identification of angiotensin-(1-7) in rat brain: evidence for differential processing of angiotensin peptides. J.Biol.Chem. 1989; 264: 16518-1652.
- Santos RAS, Silva SEAC, Maric C, Silva DM, Machado RP, Bul DI, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci. USA. 2003; 100: 8258-8263.
- Donoghue M, Hsieh F, Aronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensinI to angiotensin 1-9. Circ Res. 2000; 87: E1-E9.
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J.Biol. Chem. 2000; 275: 33238-33243.
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002; 417: 822-828.
- Santos RA. Angiotensin-(1-7). Hypertension. 2014; 63: 1138-1147.
- Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the Angiotensin converting enzyme 2-Angiotensin (1-7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol. Lausanne. 2014; 4: 201-215.
- Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am.J. Physiol Heart Circ. Physiol. 2016; 310: H137-H152.
- Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin-12, a possible component of the reninangiotensin system. Biochem Biophys Res Comm. 2006; 350: 1026-1031.
- Westwood BM, Chappell MC. Divergent pathways for the angiotensin-(1-12) metabolism in the rat circulation and kidney. Peptides. 2012; 35:190-195.
- Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa- Fraga F, et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res. 2013; 112: 1104-1111.
- Garabelli PJ, Modrall JG, Penninger JM, Ferrario CM, Chappell MC. Distinct roles for angiotensin converting enzyme 2 and carboxypeptidase A in the processing of angiotensins in the murine heart. Exp Physiol. 2008; 96: 613- 621.
- Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, et al. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme homologue ACE2 Circulation. 2003; 108: 1707-1712.
- Prada HJA, Ferreira AJ, Katovich MJ, Shenoy V, Qi Y, Santos, RA, et al. Structure-basedidentification of small-molecule angiotensin-converting enzyme 2 activators asnovel antihypertensive agents. Hypertension. 2008; 51: 1312-1317.
- De Maria ML, Araujo LD, Fraga-Silva RA, Pereira LA, Ribeiro HJ, Menezes GB, et al. Anti-hypertensive effects of diminazeneaceturate: an angiotensinconverting enzyme 2 activator in rats. Protein Pept Lett. 2016; 23: 9-16.
- Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, et al. The angiotensin-convertingenzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection againstlung fibrosis and pulmonary hypertension. Is J Respir Crit Care Med. 2010; 182: 1065-1072.
- Qi Y, Zhang J, Cole-Jeffrey CT, Shenoy V, Espejo A, Hanna M, et al. Diminazene aceturate enhances angiotensinconverting enzyme 2 activity and attenuates ischemia-induced cardiac pathophysiology. Hypertension. 2013; 62: 746-752.
- Haber PK, Ye M, Wysocki J, Maier C, Haque SK, Batlle D. Angiotensinc onverting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: studies in vivo, ex vivo, and in vitro. Hypertension. 2014; 63: 774-782.
- Raffai G, Khang G, Vanhoutte PM. Angiotensin-(1-7) augments endothelium dependent relaxations of porcine coronary arteries to bradykinin by inhibiting angiotensin-converting enzyme 1. J Cardiovasc Pharmacol. 2014; 63: 453- 460.
- Velkoska E, Dean RG, Burchill L, Levidiotis V, Burrell LM. Reduction in renal ACE2 expression in subtotal nephrectomy in rats is ameliorated with ACE inhibition.Clin Sci (Lond). 2010; 118: 269-279.
- Poglitsch M, Domenig O, Schwager C, Stranner S, Peball B, Janzek E, et al. Recombinant Expression and Characterization of Human and Murine ACE2: Species-Specific Activation of the Alternative Renin-Angiotensin-System. Int J Hypertens. 2012; 2012: 428950.
- Haschke M, Schuster M, Poglitsch M, Loibner H, Salzberg M, Bruggisser M, et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013; 52: 783-792.
- Hao PP, Yang JM, Zhang MX, Zhang K, Chen YG, Zhang C, et al. Angiotensin-(1-7) treatment mitigates right ventricular fibrosis as a distinctive feature of diabetic cardiomyopathy. Am.J.Physiol Heart Circ. Physiol. 2015; 308: H1007-H1019.
- Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J, et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2013; 59: 529-538.
- Bae EH, Fand F, William VR, Konvalink A, Zhou X, Patel VB, et al. Murine recombinant ACE2 attenuates kidney injury in experimental Alport syndrome. Kid Int. 2017; 91: 1347-1361.
- WysockiJ YEM, Khattab AM, Fogo A, Marin A, Kanwar DNVY, Osborn M, et al. ACE2 amplification limited to the circulation does not protect mice from development of diabetic neprhopathy. Kid Iint. 2017; 91: 1336-1346.
- Wiemer G, Dobrucki LW, Louka FR, Malinksi T, Heitsch H. AVE 0991, a nonpeptide iico fof hte effects of Angiotenisn-(1-7) on the endothelium. Hypertension. 2002; 40: 847-853.
- Santos RA, Ferreira AJ. Pharmacological effects of AVE 0991: a nonpepditde angiotensin-(1-7) receptor agonist. Cardiovasc Drug Rev. 2006; 24: 239-246.
- Marques FD, Ferreira AJ, Sinisterra RD, jacoby BA, Sousa FB, Caliari MV, et al. An oral formulation of Ang-(1-7) produces carioproteciv eeffecs in in infarcted andi soproteeronaol trated rats. Hypertension. 2011; 57: 477-483.
- Kluskens LD, Nelemans SA, Rink R, deVries L, Meter-Arkema a, Wang Y. Angiotensin-(1-7) with Thioether Bridge: An Angiotensin-Converting Enzyme- Resistant, Potent Angiotensin-(1-7) Analog J Pharm Exp Therap. 2009; 328: 849-854.
- Durik M, Veghel VR, Kuiper A, Rink R, Haas M, Kaanbi J, et al. The effect of the thioether-bridged, stabilized Angiotensin-(1-7) analogue cyclic Ang- (1-7) on cardiac remodeling and endothelial function in rats with myocardial infarction. Int J Hypertension. 2012; 2012: 536426.
- Webster A, Devocelle M, Tallant EA, Chappell MC, Gallagher PE, Paradisi F. Stabilization of Angiotensin-(1-7) by key substitution with a cyclic non-natural amino acid. Amino Acids. Epub. 2017; 125.
- Cruz-Diaz N, Wilson BA, Pirro NT, Brosnihan KB, Marshall AC, Chappell MC. Identification of dipeptidyl peptidase 3 as the Angiotensin-(1-7) degrading peptidase in human HK-2 renal epithelial cells. Peptides. 2016; 83: 29-37.
- Wilson BA, Cruz-Diaz N, Marshall AC, Pirro NT, Su Y, Gwathmey TM, et al. An angiotensin-(1-7) peptidase in the kidney cortex, proximal tubules, and human HK-2 epithelial cells that is distinct from insulin degrading enzyme. Am J Physiol Renal Physiol. 2015; 308: F594-F601.
- Bruce E, Shenoy V, Rathinasabapathy A, Espejo A, Horowitz A, Oswalt A, et al. Selective activation of angiotensin AT2 receptors attenuates progression of pulmonary hypertension and inhibits cardiopulmonary fibrosis.Br J Pharm. 2015; 179: 2219-2231.
- Leonhardt J, Villela DC, Teichmann A, Münter LM, Mayer MC, Mardahl M, et al. Evidence for Heterodimerization and Functional Interaction of the Angiotensin Type 2 Receptor and the Receptor MAS. Hypertension. 2017; 69:1128-1135.
- Yousif MH, Benter IF, Diz DI, Chappell MC. Angiotensin-(1-7)-dependent vasorelaxation of the renal artery exhibits unique angiotensin and bradykinin receptor selectivity. Peptides. 2017; 90: 10-16.
- Gwathmey TM, Alzayadneh EM, Pendergrass KD, Chappell MC. Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am J Physiol Regul Integr Comp Physiol. 2012; 302: R518-R530.
- Ellis B, Li XC, Miguel-Qin E, Gu V, Zhuo JL. Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am J Physiol Regul Integr Comp Physiol. 2012; 302: R494-R509.
- Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine reninangiotensin system. Clin Sci (Lond). 2012; 123: 273-284.
- Cook JL, Re RN. Lessons from in vitro studies and a related intracellular angiotensin II transgenic mouse model. Am J Physiol Regul Integr Comp Physiol. 2012; 302: R482-R493.