
Abstract
The main objective of selecting this topic because of many pharmaceutical companies receives the 483 observations/ warning letters from the USFDA. The reason of most of 483 observations/warning letters was inadequate investigation of Laboratory Non-Conformances. It includes Out of specification/Out of trend investigation. The inadequate investigation because of unawareness of regulatory guidance requirements while performing the investigation. If any company receives 483 observations/warning letter it will impact on company reputation in the market and also impact on company revenue. So as to avoid this we should know the some basic concept while performing the out of specification/Out of trend investigation.
Keywords: Applicability; Definitions; Phase I (Initial Laboratory Investigation); Phase II (Full Scale OOS investigation); Manufacturing investigation hypothesis testing/experimentation; Re-sampling; Retesting; Inconclusive OOS; Key point to be considered while performing OOS investigation; Conclusion; USFDA Observation on OOS investigation
Introduction
As per current good manufacturing practice for finished pharmaceuticals. Subpart J--Records and Reports (211.192 Production record reviews). All drug product production and control records, including those for packaging and labeling, shall be reviewed and approved by the quality control unit to determine compliance with all established, approved written procedures before a batch is released or distributed [1]. Any unexplained discrepancy (including a percentage of theoretical yield exceeding the maximum or minimum percentages established in master production and control records) or the failure of a batch or any of its components to meet any of its specifications shall be thoroughly investigated, whether or not the batch has already been distributed [2]. The investigation shall extend to other batches of the same drug product and other drug products that may have been associated with the specific failure or discrepancy [3-5]. A written record of the investigation shall be made and shall include the conclusions and follow-up. Even if a batch is rejected based on an OOS result, the investigation is necessary to determine if the result is associated with other batches of the same drug product or other products. Batch rejection does not negate the need to perform the investigation. The investigation should be thorough, timely, unbiased, well-documented, and scientifically sound [6].
Applicability: OOS/OOT/Atypical Results Investigation Applicable To
• Batch release testing and testing of starting materials.
• In-Process Control testing: if data is used for batch calculations/decisions and if in a dossier and on Certificates of Analysis.
• Stability studies on marketed batches of finished products and or active pharmaceutical ingredients, on-going/follow up stability (no stress tests) 83 observation/warning letters.
• Previous released batch used as reference sample in an OOS investigation showing OOS or suspect results.
• Pharmacopoeias have specific criteria for additional analyses of specific tests (i.e. dissolution level specification for S1, S2 & S3 testing; Uniformity of dosage unit’s specification for testing of 20 additional units; Sterility Testing). However if the sample test criteria is usually the first level of testing and a sample has to be tested to the next level this should be investigated as it is not following the normal trend.
• Batches for clinical trials.
OOS Investigation NOT Applicable To
• In-process tests that are performed for the purpose of monitoring and/or adjusting the process (e.g. pH, viscosity).
• The studies conducted at variable parameters to check the impact of drift (e.g. process validation at variable parameters).
• Stress tests conducted on sample ex. Temperature excursion study, photo stability study.
Important Definitions
1) Out-of-Specification (OOS) Result: Test results that fall outside of established acceptance criteria which have been established in official compendia and/or by company documentation (i.e., Raw Material Specifications, In-Process/Final Product Testing, etc.).
2) Atypical/Aberrant/Anomalous Result: Results that are still within specification but are unexpected, questionable, irregular, deviant or abnormal. Examples would be chromatograms that show unexpected peaks, unexpected results for stability test point, etc.
3) Obvious Error: Investigation is to determine whether there has been a clear obvious error due to external circumstances such as power failure or those that the analyst has detected prior to generating data such as spilling sample.
Examples:
Calculation error: Analyst and supervisor to review both initial and date correction.
Power outage: Analyst and supervisor document the event, annotate “power failure; analysis to be repeated” on all associated analytical documentation.
Equipment failure: Analyst and supervisor document the event, annotate “equipment failure; analysis to be repeated” cross reference the maintenance record.
Testing errors: for example, spilling of the sample solution, incomplete transfer of a sample; the analyst must document immediately. For microbiology it could be growth on a plate not in the test sample area, negative or positive controls failing.
Incorrect instrument parameters: For example setting the detector at the wrong wavelength, analyst and supervisor document the event, annotate “incorrect instrument parameter”; analysis to be repeated” on all associated analytical documentation.
4) Assignable cause: An identified reason for obtaining an OOS or aberrant/anomalous result.
5) No assignable cause: When no reason could be identified.
6) Invalidated test: A test is considered invalid when the investigation has determined the assignable cause.
7) Reportable result – The final analytical result. This result is appropriately defined in the written approved test method and derived from one full execution of that method, starting from the original sample.
8) Hypothesis/Investigative testing: Testing performed to help confirm or discount a possible root cause i.e. what might have happened that can be tested: - for example it may include further testing regarding sample filtration, sonication /extraction; and potential equipment failures etc. Multiple hypothesis can be explored
9) Re-test: Performing the test over again using material from the original sample composite, if it has not been compromised and/or is still available. If not, a new sample will be used.
10) Re-sample: A new sample from the original container where possible, required in the event of insufficient material remaining from original sample composite or proven issue with original sample integrity.
11) Most probable cause: Scientifically justified determination that the result appears to be laboratory error.
12) Corrective action: Action to eliminate the cause of a detected non-conformity or other undesirable situation. Note: Corrective action is taken to prevent recurrence whereas preventive action is taken to prevent occurrence.
13) Preventative action: Action to eliminate the cause of a potential non-conformity or other undesirable potential situation.
Note: Preventive action is taken to prevent occurrence whereas corrective action is taken to prevent recurrence.
Phase I: Investigation (Initial Laboratory Investigation)
The phase I investigation involves the assessment of laboratory data and Verifications of initial preparation. This activities performed by analyst and supervisor. Whenever possible, this should be done before test preparations (including the composite or the homogenous source of the aliquot tested) are discarded. This way, hypotheses regarding laboratory error or instrument malfunctions can be tested using the same test preparations. Examination of the retained solutions should be performed as part of the laboratory investigation [7,8].
• Re-injection of same solution ---To rule out the error related to instrument malfunctioning.
• Re-dilution or Re-pipetting of same solution----To rule out the error related to dilution or pipetting.
It should not be assumed that OOS test results are attributable to analytical error without performing and documenting an investigation.
If cause of out of specification is identified then retesting or recalculation to be performed. If results found within the limit, initial data to be invalidated. Retesting or recalculated data to be consider for final reporting and release.
For better understanding of phase-I investigation refer flow diagram-A (Figure 1).
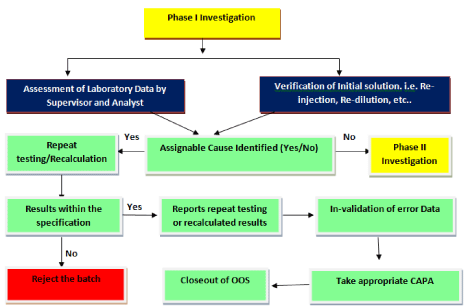
Figure 1: Flow diagram- A (Phase I Investigation).
When the initial assessment does not determine that laboratory error caused the OOS result and testing results appear to be accurate, a full-scale OOS investigation using a predefined procedure should be conducted [7].
Phase II: Investigation (Full Scale OOS Investigation)
Phase II investigation involves:
• Production process review/ Manufacturing Investigation
• Hypothesis testing/Experimentation
• Additional laboratory testing
Production process review/manufacturing Investigation
When the initial laboratory investigation does not determine the cause of OOS results, full-scale OOS investigation using a predefined procedure should be conducted. This investigation may consist of a production process review and/or additional laboratory work. The objective of such an investigation should be to identify the root cause of the OOS result and take appropriate corrective and preventative action. A full-scale investigation should include a review of production and sampling procedures, and will often include additional laboratory testing. A full-scale OOS investigation should consist of a timely, thorough, and well-documented review [8].
If this part of the OOS investigation confirms the OOS result and is successful in identifying its root cause, the OOS investigation may be terminated and the product rejected. However, a failure investigation that extends to other batches or products that may have been associated with the specific failure must be completed.
Hypothesis testing/experimentation
Hypothesis/Investigative testing: Description of the testing should be written, and then approved by QA prior to initiating investigational testing. The requirements of investigational testing listed below:
The description must fully document;
• The hypothesis to the test the root cause being investigated.
• What samples will be tested.
• The exact execution of the testing.
• How the data will be evaluated
On completion of the Analyst and Supervisor investigation hypothesis testing can be started. Hypothesis testing performed to help confirm or discount a possible root cause i.e. what might have happened that can be tested. Hypothesis testing applicable to Phase I and Phase II. The initial hypothesis testing can include the original working stock solutions but should not include another preparation from the original sample. The initial hypothesis testing can involve re-measurement of the original preparation or working solutions, however retesting is when the original sample or composite sample is used to perform analysis [9].
Example:
It may include further testing regarding sample filtration, Sonication /extraction; and potential equipment failures etc.
• Original Solutions can be re-injected as part of an investigation where a transient equipment malfunction is suspected. Re-injections can provide strong evidence that the problem should be attributed to the instrument, rather than the sample or its preparation.
• Re-dilution from original solution in case of multiple dilutions to find out dilution error.
• Re-extraction of a dosage unit, where possible, can be performed to determine whether it was fully extracted during the original analysis.
Investigational testing may not be used to replace original suspect analytical results. It may only be used to confirm or discount a probable cause.
Additional laboratory testing
Phase II investigation may includes additional laboratory testing.
These include:
• Re-sampling and
• Retesting a portion of the original sample.
Criteria for Re-sampling: It involves the collecting a new sample from the batch.
• If insufficient quantity of the original sample remains to perform all further testing then the procedure for obtaining a resample must be discussed and agreed by QA/Contract Giver/QA equivalent. The process of obtaining the resample should be recorded within the laboratory investigation.
• Re-sampling should be performed by the same qualified, validated methods that were used for the initial sample.
• Sound scientific justification must be employed if resampling is to occur.
• An investigation might conclude that the original sample was prepared improperly and was therefore not representative of the batch quality.
• If the investigation determines that the initial sampling method was inherently inadequate, a new accurate sampling method must be developed, documented, and reviewed and approved by the QCU.
• If the initial sample given to the laboratory for analysis has become in some way adulterated (e.g., through breakage or exposure to heat, light, or moisture) or was exhausted in the testing process, it may be acceptable to resample the batch under the same constraints as described earlier.
• Control mechanisms for examination of additional specimens should be in accordance with predetermined procedures and sampling strategies (§ 211.165(c)).
Criteria for retesting:
• The decision to retest should be based on sound scientific judgment. The test plan must be approved before re testing occurs.
• Retesting Performed on Original sample.
• Can be a 2nd aliquot from the same sample that was the source of the original failure.
• The sample used for the retesting should be taken from the same homogeneous material that was originally collected from the lot, tested, and yielded the OOS results. For a Liquid it may be from the original unit liquid product or composite of the liquid product and for a solid, it may be an additional weighing from the same sample composite prepared for the original test.
• Retests performed by an analyst other than the one who performed the original test. (It’s a part of retesting Plan).
• A second analyst performing a retest should be at least as experienced and qualified in the method as the original analyst.
• The minimum number of retests should be documented within the procedure and be based upon scientifically sound principles. Any statistical review with regards to %RSD and repeatability should relate to the values obtained during method validation (accuracy, precision, and intermediate precision). The number of retests should be statistically valid; papers have suggested 5, 7, or 9.
Flow diagram- B (Phase-II Investigation).
Handling of Inconclusive OOS
Inconclusive OOS, Product /Material can be released by verifying following Key points (Figure 2):
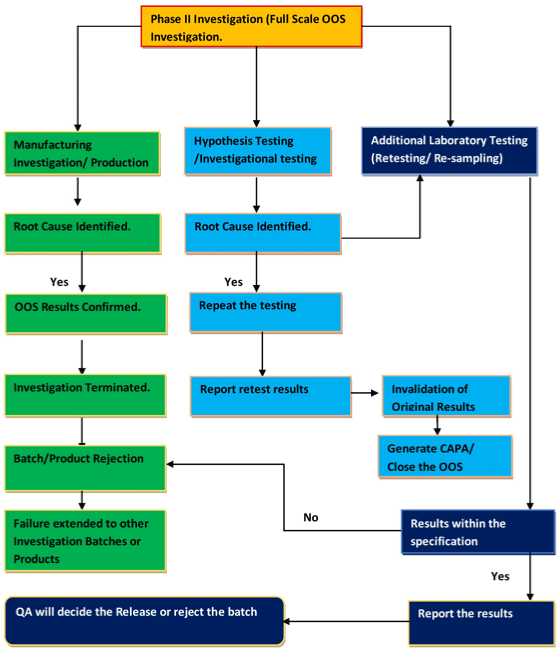
Figure 2: Flow diagram- B (Phase-II Investigation).
• A comprehensive laboratory investigation (Phase 1) fails to reveal any laboratory error.
• Review of events during production of the batch reveals no aberrations or indication of unusual process variation.
• Review of the manufacturing process and product history demonstrates that the process is robust.
• The Six/seven passing retest results are all well within the known limits of variability of the method used.
• Batch results from in-process monitoring, content uniformity, dissolution, and other tests are consistent with the passing retest results.
After a comprehensive investigation, Quality control might conclude that the initial OOS result did not reflect the true quality of the batch. Any decision to release a batch, in spite of an initial OOS result that has not been invalidated, should come only after a full investigation has shown that the OOS result does not reflect the quality of the batch. In making such a decision, Quality Assurance/ QP should always err on the side of caution [10].
Key Point to be Consider during OOS Investigation
• Initial hypothesis testing can include the original working stock solutions but should not include another preparation from the original sample.
• Even if a batch is rejected based on an OOS result, the investigation is necessary to determine if the result is associated with other batches of the same drug product or other products and identification and implementation of corrective and preventative action. Batch rejection does not negate the need to perform the investigation.
• It is important when considering performing additional testing that it is performed using a predefined retesting plan to include retests performed by an analyst other than the one who performed the original test. A second analyst performing a retest should be at least as experienced and qualified in the method as the original analyst.
• FDA inspections have revealed that some firms use a strategy of repeated testing until a passing result is obtained, then disregarding the OOS results without scientific justification. This practice of “testing into compliance” is unscientific and objectionable under CGMPs.
• The firm’s predetermined retesting procedures should contain a point at which the additional testing ends and the batch is evaluated.
• Failure investigation that extends to other batches or products that may have been associated with the specific failure must be completed.
• Investigational testing/Hypothesis testing may not be used to replace an original suspect analytical result. It may only be used to confirm or discount a probable cause.
• This Hypothesis testing may continue from the remeasurement of the original preparations.
• Once a batch has been rejected there is no limit to further testing to determine the cause of failure, so that corrective action can be taken.
• The decision to reject cannot be reversed as a result of further testing.
• If the investigation determines that the initial sampling method was inherently inadequate, a new accurate sampling method must be developed, documented, and reviewed and approved by the Quality Assurance responsible for release. A consideration should be given to other lots sampled by the same method.
• Any decision to release a batch, in spite of an initial OOS result that has not been invalidated, should come only after a full investigation has shown that the OOS result does not reflect the quality of the batch. In making such a decision, Quality Assurance/ QP should always err on the side of caution.
• Products that are the subject of approved full and abbreviated new drug applications, regulations require submitting within 3 working days a Field Alert Report (FAR) of information concerning any failure of a distributed batch to meet any of the specifications established in an application.
Conclusion
• If no laboratory or calculation errors are identified in the Phase I and Phase II there is no scientific basis for invalidating initial OOS results in favour of passing retest results. All test results, both passing and suspect, should be reported (in all QC documents and any Certificates of Analysis) and all data has to be considered in batch release decisions.
• When clear evidence of laboratory error exists, laboratory testing results should be invalidated. The firm should determine the source of that error and take corrective action to prevent recurrence.
• When evidence of laboratory error remains unclear, a fullscale OOS investigation should be conducted by the manufacturing firm to determine what caused the unexpected results (FDA).
• An initial OOS result does not necessarily mean the subject batch fails and must be rejected. The OOS result should be investigated, and the findings of the investigation, including retest results, should be interpreted to evaluate the batch and reach a decision regarding release or rejection which should be fully documented.
• In those cases where the investigation indicates an OOS result is caused by a factor affecting the batch quality (i.e., an OOS result is confirmed), the result should be used in evaluating the quality of the batch or lot. A confirmed OOS result indicates that the batch does not meet established standards or specifications and should result in the batch’s rejection, in accordance with § 211.165(f), and proper disposition. Other lots should be reviewed to assess impact.
• In case of confirmed OOS, the investigation changes from an OOS investigation into a batch failure investigation, which must be extended to other batches or products that may have been associated with the specific failure (§ 211.192).
• If the investigation determines that the initial sampling method was inherently inadequate, a new accurate sampling method must be developed, documented, and reviewed and approved by the Quality Assurance responsible for release. A consideration should be given to other lots sampled by the same method.
• Investigational/Hypothesis testing may not be used to replace an original suspect analytical result. It may only be used to confirm or discount a probable cause.
USFDA Observations on OOS Investigation
Observation 1: Invalidated Out-Of-Specification (OOS) results without adequate investigation and scientific justification. Example: Obtained OOS results for the impurity during stability testing of injection batches. OOS investigation reports stated that the postulated cause was “poor column efficiency”, although no chromatographic abnormalities were noted and system suitability criteria were met. During the inspection, lab management indicated that retention times, theoretical plates, and tailing factor appeared appropriate and no specific root cause had been demonstrated. Repeated the analyses, obtained passing results, and invalidated the OOS results.
Observation 2: OOS investigation of the failure of to meet the specifications under accelerated stability conditions. While the investigation lacked a demonstrated assignable root cause in the laboratory, obtained passing results during repeat analysis and invalidated the OOS without a Phase II production investigation.
Observation 3: Investigations of Out-Of-Specification (OOS) results were inadequate. For example, in multiple instances, you disregarded the original failing result based on a retest, but you lacked a Phase 1 laboratory investigation to support invalidation of the result. You also often lacked Phase 2 investigations to evaluate your manufacturing operation for potential root causes.
Observation 4: Investigated numerous OOS results as “incidents” and not as OOS results.
Observation 5: Failed to thoroughly investigate Out-of- Specification (OOS) assay test. Retested the samples and invalidated the OOS results without any scientific justification and released these lots into the U.S. market (21 CFR 211.192).
Observation 6: You firm failed to establish and follow adequate written procedures for cleaning and maintenance of equipment (21 CFR 211.67(b)). The cleaning validation for your non-dedicated tank, used to manufacture your drug product was inadequate. Your High- Performance Liquid Chromatography (HPLC) chromatograms for residual disinfectant showed significant peaks for rinse samples with a retention time similar to that of your cleaning agent. You failed to investigate these peaks. During the inspection, you integrated these peaks, which yielded OOS results for residual disinfectant. Your cleaning validation failed multiple rinse samples tested for residual disinfectant.
Observation 7: Failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed (21 CFR 211.192). Your original atomic absorption analysis of sample was Out-Of Specification (OOS). A retest of the sample was also OOS. A third sample was retested and found within specifications. You invalidated the OOS results without justification or documented investigation.
Observation 8: Lacked in thorough investigations into root causes, and failed to implement prompt and effective Corrective Actions and Preventive Actions (CAPA).
Observation 9: Failed to establish adequate written responsibilities and procedures applicable to the quality control unit and to follow such written procedures (21 CFR 211.22(d)). You lacked adequate written procedures for various functions, including, but not limited to customer complaints, recalls, annual product review, outof- specification (OOS) or deviation investigations, change control, CGMP-related training, issuing batch records, documenting batch record review, cleaning, storage conditions.
Observation 10: Your firm does not ensure that complete data from testing of your API are included in the official batch record and reviewed by your quality unit. For example, you reported passing results for related substances. However, our investigator found unreported analyses including out-of-specification (OOS) results for the same lot acquired earlier on the same date, and on the next day as the reported results.
Observation 11: Lacked of adequate procedures for investigating, and scientific justification to invalidate, OOS results.
Observation 12: OOS Results for Residual Solvent You initiated investigation for an initial OOS. The investigation did not reveal laboratory testing anomalies. You tested another sample preparation three times and obtained results very close to the specification upper limit. You invalidated the initial failing result, stating that your statistical analysis showed a significant difference between the original value and the retest results. Your investigation lacked further assessment of the root cause of the failing result.
Observation 13: Out-Of-Specification (OOS) results observed for viscosity test. The next two retest values were also OOS. You failed to conduct laboratory and manufacturing investigations into these OOS results, which included identifying a root cause and implementing Corrective Actions and Preventive Actions (CAPA). Instead, you rejected the batch without conducting an adequate investigation.
Observation 14: Quality control laboratory disregarded multiple Out-Of-Specification (OOS) impurity results without justification.
Observation 15: Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed (21 CFR 211 192). Your firm frequently invalidated initial Out-Of-Specification (OOS) laboratory results without an adequate investigation that addressed potential manufacturing causes.
Observation 16: Our investigators documented that your investigations into Out-Of-Specification (OOS) test results were not thorough, timely, or based on scientific rationales. Your investigations did not adequately determine root cause. Stability Failure: Investigation of Two different batches failed stability testing. During the inspection, we reviewed your initial OOS investigation in which you determined that the stability failures were caused by an excipient used to manufacturing. In the same investigation, you also concluded, without performing a science-based health hazard evaluation, that such impurities do not pose health risks. You continued to distribute other batches of the same product while your OOS investigation remained open for more than five months.
References
- Guidance for Industry Investigating Out-Of-Specification (OOS) Test Results for Pharmaceutical Production.
- MHRA Guideline on Out of Specification. 2018.
- Reference book on Handling Laboratory and Manufacturing Deviations Robert B. Kirsch R. B. Kirsch Consulting, Arlington Heights, Illinois, USA.
- The GMP Questions & Answers Guide - GMP Advisor Version 02 of 2020.
- Guide To Inspections of Pharmaceutical Quality Control Laboratories.
- Current Good Manufacturing Practice For Finished Pharmaceuticals 21 CFR 211, 21 CFR 210.
- Guidance for Industry Q7A Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients.
- Stability testing of active pharmaceutical ingredients and finished pharmaceutical products WHO Technical Report Series, No. 1010. 2018.
- USFDA issue Warning Latter to pharmaceutical industry.
- WHO guidelines on quality risk management. WHO Technical Report Series No. 981. 2013.