
Research Article
Austin J Pharmacol Ther. 2022; 10(1).1159.
Antibacterial and Antioxidant Properties of Endophytic Fungi Extracts from Cola acuminata (Sterculiaceae)
Hzounda Fokou JB1,2*, Lunga PK2, Momo ER2, Manaoda AV3, Yimgang LV2, Mbetyoumoun HM2, Lazar IM4 and Boyom FF2
1Laboratory for Pharmacology, Department of Pharmaceutical Sciences, University of Douala, Douala, Cameroon
2Anti-Microbial Agents Unit, Laboratory for Phytobiochemistry and Medicinal Plants Studies, Department of Biochemistry, University of Yaoundé I, PO Box 812, Yaoundé, Cameroon
3Laboratory of Analytical Chemistry and Toxicology, Department of Pharmaceutical Sciences, University of Douala, Douala, Cameroon
4Faculty of Psychology and Educational Sciences, University of Bucharest, Bucharest, Romania
*Corresponding author: Jean Baptiste Hzounda Fokou, Laboratory for Pharmacology, Department of Pharmaceutical Sciences, University of Douala, PO Box 2701, Douala, Cameroon
Received: January 22, 2022; Accepted: February 17, 2022; Published: February 24, 2022
Abstract
Currently, the disease burden from pneumonia remains a major public health problem. In this regard, exploring endophytic fungal extracts from traditionally used plants could be a promising approach in this light. Therefore, this work was conceived with the aim of evaluating the antibacterial and antioxidant activities of two endophytic extracts of Cola acuminata against pneumoniacausing bacteria. The identification led to the acquisition of two endophytic fungi: Trichoderma harzianum and Trichoderma afroharzianum. From ten (10) extracts, two (CAB31P1 and CAF71N2) were active on the tested bacteria, and their MICs ranged from 12.5 to 100 μg/ml. CAF71N2 displayed better antioxidant activity with IC50 values of 74.75, 12.70 and 5.66 μg/ml for the reducing power of Fe3+, NO and OH radical scavenging capacities, respectively. The extracts revealed no cytotoxicity (CC50>100 μg/ml) on the two cell lines tested. These results suggest that the endophytic extracts from Cola acuminata could serve as a source for the isolation of potent antibacterial and antioxidant compounds for the treatment of pneumonia.
Keywords: Cola acuminata; Endophytic fungi; Antibacterial; Antioxidant; Pneumonia
Introduction
Pneumonia is a disease resulting from a complex set of processes, starting with contact with an infective microorganism and culminating in invasion of the lower respiratory system [1]. Infective microorganisms such as Streptococcus pneumoniae, Haemophilus influenzae, Klebsiella pneumoniae, Staphylococcus aureus and Escherichia coli are the most incriminated bacteria of this infection [1-3]. The disease burden from pneumonia is a current threat to humanity, causing greater deaths and economic repercussions than any other cause of disease, and it is one of the leading causes of death [1-3]. Worldwide, pneumonia remains the deadliest communicable disease, causing 3.0 million deaths worldwide [4]. In Cameroon, pneumonia was the third leading cause of death, accounting for 22.3% of cases in a study by Tazinya et al. [5] in Bamenda, while 15% and 31% of cases were recorded in Buea for children and post-neonatal deaths, respectively [6].
Once these bacteria reach the alveolar space, macrophages engulf them and trigger signal molecules such as cytokines that recruit other inflammatory cells, such as neutrophils, to the site of infection [7]. One of the effects of the former is the overproduction of reactive oxygen species (ROS) and reactive nitrogen species (RNS), such as Fe3+, OH⋅-, ONOO⋅-, HClO, H2O2, and NO, which leads to oxidative stress. The resulting effect of these actions is inflammation of the lung parenchyma, which makes the lining capillaries “leaky”. Consequently, the alveolar sacs are filled with pus and fluid, thereby making breathing difficult and limiting oxygen, leading to pneumonia [2,8].
Disease management measures primarily rely on chemotherapy using antibiotics such as those from fluoroquinolone and tetracycline classes. Despite their effectiveness, the latter face several secondary effects (sore throat, nausea, diarrhea), cost and resistance [9]. Hence, discovering new alternative agents is indispensable for the management of this disease.
The use of medicinal plants remains an alternative source of therapies since it contains substances that can be used for therapeutic purposes or that are precursors for the synthesis of useful drugs. Indeed, for millennia, several infections are being treated with medicinal plants [10,11]. Endophytes, microorganisms associated with living plant tissues that produce no apparent indication of their presence in the plant and seem not to cause harm to the host [12,13], are able to produce rich bioactive compounds with a high level of structural diversity, conferring interesting biological activities to them [14-16].
Cola acuminata is a slender tree found all over the western cost of Africa. It is used mostly for their economic aspects as well as traditionally and has displayed antimicrobial activity against Bacillus subtilis, Staphylococcus aureus, Escherichia coli, and Klebsiella pneumonia [17]. Endophytic fungi inhabiting Cola acuminata have revealed antifungal activities against some multi-resistant Candida species [18], although their antibacterial activity remains untapped. As a result, this work design with the aim to evaluate the antibacterial and antioxidant potentials of crude extracts from two endophytic fungi of Cola acuminata.
Materials and Methods
Materials
The fungal material was made of two endophytic fungi isolated from the branch (CAB31) and leaf (CAF71) of Cola acuminata. Stored at -80°C in 50% glycerol at the antimicrobial and biocontrol agent units. Bacterial strains, including Staphylococcus aureus ATCC43300, Streptococcus pneumoniae hm145 and isolates, were used for antibacterial assays. The isolates of Klebsiella pneumoniae and Escherichia coli were obtained from Centre Pasteur of Cameroon. Vero (ATCC CRL 1586) cells were equally obtained from Centre Pasteur of Cameroon, and RAW 264.7 (ATCC #TIB-71) cells were obtained from the Noguchi Memorial Institute for Medical Research, Ghana. The culture media were potato dextrose broth (HIMEDIA) and nutrient broth (LyophiChem). DPPH was from SIGMA.
Methodology
Identification of species
The identification of the two species was performed using Fourier transform infrared-attenuated total reflection (FTIR-ATR) spectroscopy by the method formally described [19] with slight modifications.
Sample preparation: This consisted of scraping mycelia from solid Leonian's medium after 4 days of growth. The sample was placed onto the ATR crystal (Golden Gate Single Reflection Diamond ATR). The infrared light passed through the crystal and interacted with the sample, which was pressed unto the crystal. Before each fungal sample scan, the device was cleaned with 70% ethanol, and after drying, the background air was taken.
Seven well-characterized fungal strains representing one fungal genus (Aspergillus sp., Trichoderma sp. and Fusarium sp.) obtained from the mycological strain collection of the antimicrobial and biocontrol agent unit (University of Yaoundé 1) were used in this study as a library for comparison purposes.
Spectral acquisition [19]: From this, different spectra were obtained showing the specific characteristics of each sample. Spectral acquisition of the strains was performed on a Tensor 27 Fourier transformed infrared spectrophotometer. The environmental conditions were maintained constant (temperature at 25°C and humidity 30%). The spectra were recorded from 4000 to 550 cm-1 with a resolution of 4 cm-1 and 260 scans for the sample and background. The scan velocity was 10 kHz, and the interferogram size was 14,220 points. The raw signal obtained was then Fourier transformed to produce a more familiar IR representation of intensity as a function of wave number. Hence, then the name ‘FT-IR’. The spectra were acquired and manipulated with Origin Pro software for Windows.
Chemometry [19]: The spectra in each region were baseline corrected by applying the rubber band method, normalized separately using the vector normalization method and then offset corrected using Origin Pro software for win cluster analysis. Principal component analysis (PCA) and linear discriminant analysis (LDA) were applied to compare samples and group the spectra with the same degree of similarity. This method calculated the Euclidean distances between all the data sets by using Ward’s algorithm method. The merging process was presented in a dendrogram regrouping the different spectra in clusters according to a heterogeneity scale.
Culture of endophytic fungi and extraction of metabolites
Cultivation of endophytic fungi for the production of secondary metabolites: The endophytic extract was prepared according to previous methodology [18,20,21] with few modifications. Briefly, the two species were grown on freshly prepared PDA plates for 4 to 7 days depending on the species. They were introduced in quadruplets into flasks containing 250 ml of nutrient broth (NB) medium and potato dextrose broth (PBD) medium. Nevertheless, only the 1-week culture was carried out for the PDB medium. The culture was then incubated at room temperature for 1, 2, 3 and 4 weeks per species, at room temperature, in the dark and with intermittent shaking. To extract the metabolites, 250 ml of ethyl acetate was added to the culture medium containing the endophytic species (after fermentation), mixed well for 10 minutes and allowed overnight until the two clear immiscible layers were formed. The upper layer of ethyl acetate containing the extract was separated using a separating funnel. The extracts were then concentrated by removing the solvents under reduced pressure at 35-40°C with a rotatory evaporator.
Determination of minimum inhibitory concentrations (MICs) of extracts against bacterial species: The MICs of extracts against bacteria were determined as described [22] using the 96-well microtiter plate format.
One hundred microliters (100 μl) of twofold diluted extracts in nutrient broth medium were introduced into the wells of the plate. Thereafter, 100 μl of the bacterial inoculum standardized at 0.5 McFarland were added to each well containing the test substances except the blank column for the sterility control. The concentrations of extracts and the positive control ranged from 3.125 to 100 μg/ml and 0.15625 to 5 μg/ml, respectively. Plates were incubated for 24 hours at 37°C, and turbidity was observed as an indication of growth. The lowest concentration inhibiting the visible growth of bacteria was recorded. Extracts with the best antibacterial activity were selected for the antioxidant and cytotoxic assays.
In vitro antioxidant and cytotoxicity assays of endophytic fungi extracts
Scavenging effect on DPPH (2,2-Diphenyl-1-picrylhydrazyl) Radical: The scavenging effect of the extracts was determined using the protocol previously described [23].
Briefly, 25 μl of extracts prepared at concentrations of 1000, 500, 250, 125 and 62.5 μg/ml was added to 75 μl of methanol solution of DPPH (0.02%) to obtain final volumes of 100 μl and final concentrations of 250, 125, 62.5, 31.25 and 15.625 μg/ml. Vitamin C prepared at an initial concentration of 1 mg/ml was used as a positive control. After 30 minutes of incubation in absolute darkness, the absorbance was read at 517 nm. Each experiment was performed in triplicate, and the percentage of inhibition of endophytic fungal extracts was calculated using the following equation: RSA = (Ao - As)/Ao × 100
where RSA: Radical Scavenging Activity; Ao: Absorbance of the blank (DPPH + methanol); As: Absorbance of DPPH Radical + endophytic fungi extract.
From %RSA, other parameters, such as the RSA50, EC50, and ARP, were deduced.
RSA50 is the concentration of extract at which 50% of the free radicals are scavenged and is obtained from a graph of %RSA as a function of the logarithmic values of extract concentrations
EC50; the efficient concentration, defined as the concentration of extract required to scavenge ½ mole of DPPH, was calculated as follows:
EC50 = RSA50/[DPPH]
ARP; Antiradical power is the inverse of the EC50. It measures the efficiency of the antiradical; hence, the larger the ARP is, the more efficient the antiradical.
ARP = 1/[EC50]
Nitric oxide radical scavenging assay: The method of Kumaresan et al. [24] with few modifications was employed to determine the nitric oxide radical scavenging activity of the extracts.
Ten microliters (10 μl) of the SNP solution was mixed with 25 μl of the extracts and vitamin C (positive control) at various concentrations ranging from 6.25 to 50 μg/ml. The mixture was incubated at 25°C. After 30 minutes, the solution was mixed with 50 μl of Griess’ reagent. The mixture was incubated at room temperature for 5 minutes, followed by the measurement of absorbance at 546 nm using a spectrophotometer (Tecan UV-1800).
Each experiment was performed in triplicate, and the radical scavenging activity (RSA) of the extracts was calculated using the following formula:
%RSA = (Ao – As)/Ao× 100
where Ao is the absorbance of the control (SNP + Griess’ reagent only) and As is the absorbance of the test samples (SNP + extract + Griess’ reagent).
From dose-response curves obtained from different concentrations of the samples, the concentration of sample required to scavenge 50% NO free radicals (50% inhibition concentration, IC50) was determined.
Hydrogen peroxide (H2O2) scavenging assay: The method described by Mukhopadhyay et al. [25] was used to determine the hydrogen peroxide (H2O2) radical scavenging activity of the samples.
To 25 μl of extract prepared at different concentrations (500, 250, 125, 62.5, and 31.25 μg/ml), 25 μl of 5 mM H2O2 was added and incubated at room temperature in the dark for 5 minutes. Thereafter, 25 μl of Fe2+ (3 mM) was added and incubated for another 5 minutes. After incubation, 75 μl of 1 mM 1,10-phenanthroline was added to the sample, homogenized and incubated for 10 minutes at room temperature. Vitamin C served as positive control. Finally, the absorbance was read at 510 nm with a spectrophotometer. The blank solution contained only Fe2+, distilled water and 1,10-phenanthroline.
Each experiment was performed in triplicate, and the hydrogen peroxide scavenging capacity of the extracts was calculated accordingly.
Scavenged H2O2 (%) = 1 -As/Ao × 100
where Ao is the absorbance of the control (Fe2+ + orthophenanthroline) and As is the absorbance of the test (Fe2+ + extract + H2O2 + ortho-phenanthroline).
From dose-response curves obtained from different concentrations of the samples, the concentration of sample required to scavenge 50% H2O2 (50% inhibition concentration, IC50) was determined.
Ferric ion reducing antioxidant power (FRAP) assay: The assay was performed according to the method described by Yefrida et al. [26] with slight modifications.
Briefly, 25 μl of each test sample was prepared at different concentrations (6.25, 12.5, 25, and 50 μg/ml) in the test plates, and 25 μl of iron (III) chloride (1.2 mg/ml) was added to the samples. Vitamin C served as positive control. Plates were incubated at room temperature for 15 minutes. After incubation, 50 μl of 1,10-phenanthroline (0.02%) was added, and then the absorbance of the mixture was determined at 510 nm through a spectrophotometer. The control contained iron (III) chloride, distilled water and 1,10-phenanthroline.
Each experiment was performed in triplicate, and the reducing capacity of the endophytic fungal extracts was calculated using the following formula:
Reducing Fe3+ (%) = 1 – As/Ao × 100
where Ao is the absorbance of the control (Fe2+ + orthophenanthroline) and As is the absorbance of the test (Fe3+ + extract + ortho-phenanthroline).
From dose-response curves obtained from different concentrations of the samples, the concentration of sample required to scavenge 50% of Fe3+ (50% inhibition concentration, IC50) was determined.
Hydroxyl radical antioxidant capacity (HORAC) assay: Hydroxyl radical scavenging activity was measured by the method of Godlewska-zylkiewicz et al. [27] with some modifications.
The reaction mixture (100 μl) contained 25 μl of (NH4)2 Fe2SO4. 6H2O (5 mM.) and 10 μl H2O2 6 mM, which was left to react to produce OH free radicals in the dark for 45 minutes. Then, 25 μl of crude extract at various concentrations (6.25 to 50 μg/mL) was added to the wells. The reaction mixture was left to further assess its scavenging activity for 60 minutes at 37°C. Thereafter, 40 μl of sodium benzoate 20 mM was added to the plates. The fluorescence was read at 400 nm with excitation at 320 nm. The blank solution contained ferrous ammonium sulfate (25 μl, 1 mM), H2O2 (10 μl), the extracts (25 μl) and distilled water (40 μl). Gallic acid served as a positive control prepared alongside the crude extracts at similar concentrations to the latter.
Each experiment was performed in triplicate, and the scavenging capacity (RSA) of the endophytic fungal extracts was calculated using the following formula.
OH RSA (%) = 1-As/Ao × 100
where Ao is the absorbance of the control without extract (hydroxyl radical + benzoic acid) and As is the absorbance of the test (hydroxyl radical + extract + benzoic acid).
From dose-response curves obtained from different concentrations of the samples, the concentration of sample required to scavenge 50% OH free radicals (50% inhibition concentration, IC50) was determined.
In vitro cytotoxicity evaluation of samples: This test was performed on Vero cells (ATCC CRL 1586) and RAW 264.7 cells (ATCC #TIB-71) using the colorimetric resazurin assay as previously described.
The test was performed in triplicate on 96-well cell culturetreated microplates as previously described. For this, 100 μl of cell suspension was introduced into the wells of 96-well plates to a final charge of 1x104 cells/well and incubated overnight at 37°C/5% CO2. After this time, the medium was removed and replaced with 96 μl of fresh medium, and 4 μl of each diluted sample was added. Plates were incubated at 37°C in a 5% CO2 incubator for 48 hours. The positive control contained podophyllotoxin (10 mM) tested at 10 μM, and the negative control wells had cells without. After this time, 10 μl of a solution of resazurin (0.15 mg/ml in PBS) was added to each well and then incubated for 4 hours. Fluorescence of the formed resazurin was measured at excitation and emission wavelengths of 530 nm and 570 nm, respectively, using an InfiniteM200 microtiter plate reader. From the resulting values of optical densities, the percentage of cell viability (CV) was calculated with Microsoft Excel software using the formula:
%CV=(At-Ab)/(At-Ac) X 100
Where,
At = Absorbance of Test, Ab= Absorbance of podophyllotoxin, Ac= Absorbance of negative control (cells).
A dose-response curve of CV against the concentration of the extracts was plotted to determine the 50% cytotoxic concentration (CC50).
Statistical analysis
The spectra were analyzed and manipulated with Origin Pro software for Windows. The data were subjected to One-way Analysis of Variance (ANOVA). Significance differences for multiple comparisons were determined when possible by the Waller-Duncan post hoc test at p≤0.05 using the Statistical Package for the Social Sciences (SPSS, version 16.0) program. Graphical evaluation was performed using Microsoft Excel 2019. RSA50 and IC50 were deducted using Microsoft Excel 2019. GraphPad Prism 5.0 software was used to determine the 50% cytotoxic concentration (CC50).
Results
Identification of species
FTIR-ATR was used for the identification of CAB31 and CAF71. In the present study, Fourier transform infrared spectroscopy was employed to differentiate between the species. Figure 1 shows the infrared absorption spectra of the two species investigated in this study. The spectra for each fungus were measured from six different isolates.
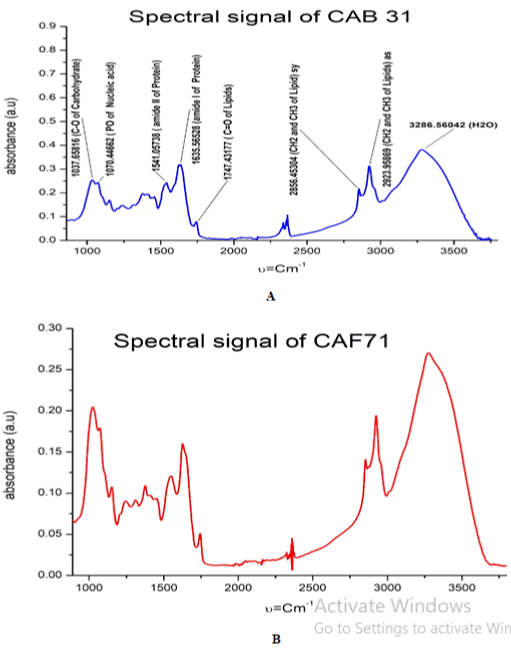
Figure 1: Spectra of CAB31 and CAF71 in the region 600-4000 cm-1, after
baseline correction and vector normalization.
To differentiate between the two species, we used principal component analysis (PCA) in selected wave number regions. To perform an accurate analysis in this wave number region, all spectra were baseline corrected and normalized. This clustering yielded a good distinction between these two species (Figure 2). Finally, the reference spectra of all isolated species in this study were compared together, and a spectral data library was established.
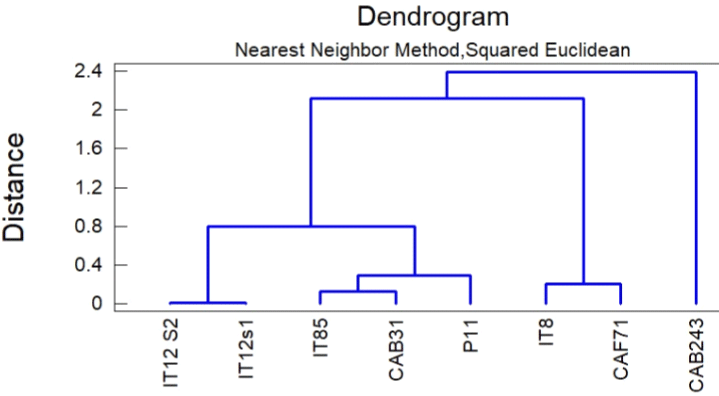
Figure 2: Dendrogram from cluster analysis of CAB31 and CAF71.
Figure 2 shows that CAB31 is in the same cluster as Trichoderma harzianum (IT85). In its part, CAF71 belongs to the same cluster as Trichoderma afroharzianum (IT8). In that regard, CAB31 and CAF71 were identified as the same species to which they are related.
In vitro antibacterial activities of endophytic fungi extracts
The screening of extracts was performed at a single concentration of 100 μg/ml, and the results are recorded in the table below.
Antibacterial screening: Ten extracts were obtained from Trichoderma harzianum and Trichoderma afroharzianum from two different culture media and four different incubation times. Table 1 shows that the endophytes produced antibacterial extracts depending on the endophyte species and the culture medium. Globally, the two extracts were active at 100 μg/ml against the above bacterial strains. Trichoderma harzianum P1 was active against all four pathogenic bacteria, while Trichoderma afroharzianum N2 was active against all bacteria except Klebsiella pneumoniae.
![]()
Sample code
Bacteria Species 100 (μg/ml)
SP
SA
KP
EC
CAB31P1
+
+
+
+
CAB31NI
-
-
-
-
CAB31N2
-
-
-
-
CAB31N3
-
-
-
-
CAB31N4
-
-
-
-
CAF71PI
-
-
-
-
CAF71NI
-
-
-
-
CAF71N3
-
-
-
-
CAF71N4
-
-
-
-
CAF71N2
+
+
-
+
+: active; -: not active; CAB31: Trichoderma harzianum; CAF71: Trichoderma afroharzianum; N: nutrient broth; P: potato dextrose broth; 1: 1-week culture; 2: 2 weeks culture; 3: 3 weeks culture; 4: 4 weeks culture; SP: Streptococcus pneumoniae; SA: Staphylococcus aureus; KP: Klebsiella pneumoniae; EC: Escherichia coli.
Table 1: The inhibitory effects of extracts at 100μg/ml on bacterial species.
Extracts showing activity on at least three bacterial species were retained for the minimum inhibitory concentration (MIC) determination. These two extracts (CAB31P1 and CAF71N2), with promising activity, were then used for the determination of their MICs.
Minimum inhibitory concentration (MIC) determination: The bacterial growth inhibition capacities of the samples were assessed, and their minimum inhibitory concentrations (MICs) are presented in the table below.
From Table 2, MIC values ranged from 12.5 to 100 μg/mL, with Trichoderma harzianum P1 showing the best activity against Streptococcus pneumoniae and Staphylococcus aureus at 25 μg/ml. Generally, the activity of Trichoderma harzianum P1 was better than that of Trichoderma afroharzianum N2 on the corresponding microorganisms.
![]()
Sample
SP
SA
KP
EC
CAB31P1
12.5
12.5
50
100
CAF71N2
50
100
/
100
Gentamicin
0.039
0.078
0.156
/
/: not determined; CAB31P1: Trichoderma harzianum 1-week culture in potato dextrose broth; CAF71N2: Trichoderma afroharzianum 2-week culture in nutrient broth; SP: Streptococcus pneumoniae; SA: Staphylococcus aureus; KP: Klebsiella pneumoniae; EC: Escherichia coli.
Table 2: Minimum inhibitory concentrations (MICs) of extracts on bacterial species.
From the results, ethyl-acetate extracts of endophytic fungi obtained from the same plant showed different levels of antimicrobial activities. These observed differences in susceptibility could be attributed to the type of isolates, nature and level of the antibacterial agents produced by each endophyte.
Antioxidant effects of promising extracts
From dose-response graphs, the 50% radical scavenging activity (RSA50) values, 50% efficient concentration (EC50) and antiradical power (ARP) were calculated, and the results are recorded in Table 3.
![]()
Sample
RSA50 (μg/ml)
EC50 (x 10-1g/mol)
ARP (x103 mol/g)
CAB31P1
687.543 ± 16.943a
13.75 ± 00a
0.072 ± 00a
CAF71N2
642.363 ± 33.657a
12.84 ± 00a
0.077 ± 00a
Vitamin C
5.496 ± 1.046b
0.001 ± 00b
1 ± 00b
Along the line, values carrying the same letter superscripts are not significantly different (P>0.05), Waller Duncan; RSA50, 50% Radical scavenging activity; EC50 = 50% Efficient Concentration; ARP = Antiradical Power; CAB31P1: Trichoderma harzianum 1-week culture in Potato dextrose broth; CAF71N2: Trichoderma afroharzianum 2-weeks culture in Nutrient Broth.
Table 3: 50% radical scavenging activity (RSA50), 50% efficient concentration (EC50) and antiradical power of DPPH radicals.
The RSA50 values of the extracts were 687.543±16.943 μg/ml and 642.363±33.657 μg/ml for Trichoderma harzianum P1 and Trichoderma afroharzianum N2, respectively. These values were much higher than that of vitamin C (5.496 ± 1.046 μg/ml), highlighting the lower effect of the extracts in comparison to the positive control. From these values, the more efficient the antioxidant capacity of the extracts, the smaller the EC50 value and the larger the ARP value. The antioxidant screening of the extracts by DPPH assay indicated the presence of antioxidant compounds in the crude extracts, although lower than that of vitamin C, which served as a positive control.
The dose-response graphs obtained were used to determine the 50% inhibitory concentration (IC50) of the samples, which are recorded in Table 4.
![]()
IC50 (μg/ml)
Sample
CAB31PI
CAF71N2
Vitamin C
Gallic Acid
NO
10.456 ± 0.401
12.703 ± 0.401
7.431 ± 0.401
-
FRAP
319.310 ± 5.597
74.756 ± 0.208
52.185 ± 1.709
-
H2O2
192.160 ± 5.070
253.170 ± 6.934
74.999 ± 13.757
-
OH
12.938 ± 1.716
5.669 ± 1.853
-
23.273 ± 0.264
Along the line, values carrying different letter superscripts are significantly different (P=0.05), Waller Duncan; IC50 = 50% inhibition concentration; CAB31P1: Trichoderma harzianum 1-week culture in potato dextrose broth; CAF71N2: Trichoderma afroharzianum 2-week culture in nutrient broth.
Table 4: 50% inhibition concentration (IC50) of samples on NO radicals.
Trichoderma harzianum P1 and Trichoderma afroharzianum N2 both revealed nitric oxide chelation effects with IC50 values of 10.456 ± 0.401 μg/ml and 12.703 ± 0.401 μg/ml, respectively. The nitric oxide activity of Trichoderma harzianum P1 was significantly higher (p≤0.05) than that of Trichoderma afroharzianum N2, both of which were significantly (p≤0.05) lower than that of vitamin C (IC50 of 4.79 ± 0.10 μg/ml), the positive control. This indicated that the extracts scavenged the nitrite radical to a lower extent than the positive controls.
Regarding the ferric reducing antioxidant power assay, the results in Table 4 clearly show that vitamin C, which served as a positive control, had the greatest reducing capacity with the lowest IC50 value of 52.185 ± 1,709 μg/ml, followed by Trichoderma afroharzianum N2 with an IC50 of 74.756 ± 0.208 μg/ml. The activity of Trichoderma afroharzianum N2 was significantly greater (p≤0.05) than that of Trichoderma harzianum P1, both of which were significantly lower (p≤0.05) in activity than vitamin C.
Regarding the hydrogen peroxide assay, the results of their IC50 values are shown in Table 4. From that table, vitamin C, which served as a positive control, had the greatest scavenging capacity with the least IC50 value of 74.999 ± 13.757 μg/ml, followed by Trichoderma afroharzianum N2 with an IC50 of 192.160 ± 5.070 μg/ml. The reducing capacities of all the samples were significantly different (p=0.05), with Trichoderma afroharzianum N2 presenting a better activity than CAB Trichoderma harzianum P1 (192.160 ± 5.070).
Table 4 shows that Trichoderma harzianum P1 and Trichoderma afroharzianum N2 possessed significantly better (p≤0.05) hydroxyl radical scavenging activities with IC50 values of 12.938 ± 1.716 μg/ ml and 5.669 ± 1.853 μg/ml, respectively, than gallic acid (23.273 ± 0.264), which served as a positive control.
Overall, although less than the reference in most cases, the antioxidant activity of Trichoderma afroharzianum N2 was significantly higher than that of Trichoderma harzianum P1 on the respective radicals.
In vitro cytotoxicity evaluation of samples
Table 5 below shows the 50% cytotoxicity concentration of samples on two cell lines.
![]()
CC50 (μg/ml)
Sample
Raw 264.7 cell
Vero cells
CAB31P1
?100
?100
CAF71N2
?100
?100
Podophyllotoxin
0.141 ± 0.014
0.177 ± 0.058
CC50 = 50% cytotoxicity concentration; CAB31P1: Cola acuminata branch after 1-week of culture in potato dextrose broth; CAF71N2: Cola acuminata leaf after 2 weeks of culture in nutrient broth.
Table 5: In Vitro Cytotoxicity Evaluation of Raw 264.7 cells and Vero cells samples.
According to the National Institute of Cancer, an extract is considered cytotoxic when it shows a CC50 value less than 20 μg/ ml. The table above presents the CC50 values of the active samples, which are all greater than 100 μg/mL. All samples were therefore not cytotoxic (CC50 values > 20 μg/ml) to either Raw or Vero cells.
Discussion
This work was designed with the aim of obtaining extracts with antibacterial and antioxidant properties from endophytic fungi inhabiting Cola acuminata. To achieve this goal, two endophytic fungi isolated from Cola acuminata were used. The identification of these strains was performed by FTIR-ATR spectroscopy. FTIRATR spectroscopy is an attractive technique for the detection and identification of pathogens. This enabled us to learn about the spectral relations between the various isolates, which in turn helped us track the propagation of the fungus and the origin of a particular isolate [19]. The present study successfully identified two Trichoderma harzianum and Trichoderma afroharzianum isolates from Cola acuminata. This goes in the straight line of what was done recently [19-21] based on the suggestion done previously [22-25]. The identified endophytic fungi T. harzianum and T. afroharzianum were then cultured with two different culture media (PDB and NB) and at different culture durations and were subjected to ethyl acetate extraction.
Antibacterial screening revealed that of the 10 extracts obtained, CAB31P1 (Trichoderma harzianum) showed activity against all four pathogenic bacterial strains, while CAF71N2 (Trichoderma afroharzianum) showed activity against three bacterial strains. No extract cultivated in NB medium for 1 week exhibited any activity; hence, this could be because NB medium below 1 week is not appropriate for the production of active principles against these pathogens. This is in accordance with what has been reported elsewhere. In fact, Vrabl et al. reported that the biomass of Trichoderma harzianum is correlated with the chemical composition of the culture medium [26]. It is known that secondary metabolite production is correlated with the chemical composition of the medium. It has been reported that when nutrients are going down, endophytes start producing active compounds against other microorganisms that can compete with them for the substrate present in the milieu [15,27,28]. Likewise, the two-week culture in NB showed activity, which could be because the fungi produced more active metabolites with adaptation during its growth phase [26]. The antibacterial activity of these two crude ethyl acetate extracts, CAB31P1 and CAF71N2, which exhibited inhibitory activities at 100 μg/mL on at least three bacterial strains, was then evaluated by determining their MICs. Hence, both extracts displayed significant activity on all bacteria, but CAB31P1 revealed the lowest MICs of 12.5 μg/mL on Streptococcus pneumoniae and Staphylococcus aureus. These results are in agreement with previous studies [16] although these are the first antibacterial results of endophytic fungi isolated from Cola acuminata.
Having unraveled the possible antibacterial activity of the extracts and characterized the relationship between their concentrations and the culture media from which they were extracted, their antioxidant potentials were equally evaluated to determine whether, in addition to their antibacterial properties, they could fight against possible oxidative stress caused by pathogenic invasion. First, in regard to Table 3, the extracts had comparable DPPH scavenging activities, which were significantly lower than that of vitamin C. CAB31P1 and CAF71N2 both revealed anti-nitric oxide activities with IC50 values of 10.456 ± 0.401 μg/ml and 12.703 ± 0.401 μg/ml, respectively, which were significantly closer to that of the positive control (Table 4). Similarly, CAF71N2 scavenged hydrogen peroxide with an IC50 of 192.160 ± 5.070 μg/ml (Table 4). To the best of our knowledge, we did not find authors who discussed the nitric oxide- and peroxidescavenging effects of endophytic extracts. CAB31P1 and CAF71N2 displayed better hydroxyl radical scavenging activities with IC50 values of 12.938 ± 1.716 μg/ml and 5.669 ± 1.853 μg/ml, respectively (Table 4), than gallic acid. Regarding the ferric reducing capacity of the extracts, vitamin C, which served as a positive control, had the greatest scavenging capacity with the lowest IC50 value of 52.185 ± 1.709 μg/ml, which was closer to that of CAF71N2 with an IC50 of 74.756 ± 0.208 μg/ml (Table 4). To the best of our knowledge, there is no report on the antioxidant effect of the Trichoderma sp extract in general and, more specifically, on the extract isolated from Cola acuminata from Cameroon. This result cannot be compared with other endophytes, as they have different synthetic abilities, and as observed in the present report, two closely related endophyte species do not have the same secondary metabolite synthetic abilities. This goes in the same line with what is established in the literature [13,18,29-31].
However, in the search for new therapeutic solutions to pathogens, the greatest worry remains the desire to find these solutions without them having undesirable side effects, such as the frequent cytotoxicity of isolated, semisynthetic or synthetic compounds. Hence, regardless of the therapeutic potential of a sample, it is necessary to evaluate its cytotoxicity. The cytotoxicity of the samples was evaluated on Vero cells and Raw 246.7 cells. Their CC50 were all greater than 100 μg/ mL. This finding is in line with the report from Damour et al., which showed that pretrichodermamide A and nafuredin isolated from trichoderma sp harbored by Cola nitida did not have cytotoxic effects on the mouse lymphoma cell line L5178Y [29].
Conclusion
In the present study, CAF71 and CAB31 were Trichoderma harzianum and Trichoderma afroharzianum, respectively. The culture of Trichoderma harzianum in potato dextrose broth for 1 week and Trichoderma afroharzianum in nutrient broth for 2 weeks displayed the best antibacterial and antioxidant effects with no cytotoxic effect on the cell lines tested. To the best of our knowledge, this is the first report on the antibacterial and antioxidant activities of endophytic fungal extracts from Cola acuminata.
Author Contribution Statement
J.B.H. F, E.R.M and L.V.Y. performed the experiments, J.B.H.F., P.K.L, E.R.M, A.V.M., L.V.Y., contributed samples/materials/ analyzed the data and drafted the article. P, M.R., A.V.M., conceived and designed the experiments. All the authors critically revised the work.
References
- Torres A & Cill C. Clinical Management of Bacterial Pneumonia (M. Cahill, Ed.). New Jersey: Springer New York. 2015.
- Torres A, Chalmers JD, Dela Cruz CS, Dominedò C, Kollef M, Martin-Loeches I, et al. Challenges in severe community-acquired pneumonia: a point-of-view review. Intensive Care Medicine. 2019; 45: 159-171.
- Torres A, Lee N, Cilloniz C, Vila J & Van Der Eerden M. Laboratory diagnosis of pneumonia in the molecular age. European Respiratory Journal. 2016; 48: 1764-1778.
- WHO. Deaths by Cause, Age, Sex, by Country and by Region, 2000- 2016. Geneva, World Health Organization. 2018: 1.
- Tazinya AA, Halle-Ekane GE, Mbuagbaw LT, Abanda M, Atashili J & Obama MT. Risk factors for acute respiratory infections in children under five years attending the Bamenda Regional Hospital in Cameroon. BMC Pulmonary Medicine. 2018; 18: 1-8.
- Agborndip E, Kadia BM, Ekaney DSM, Mbuagbaw LT, Obama MT, et al. Under-Five Mortality in Buea Health District, Southwest Cameroon: Evidence from a Community-Based Birth Cohort Study of Rate, Causes, and Age- Specific Patterns. International Journal of Pediatrics (United Kingdom). 2020.
- Deng X, Luyendyk JP, Ganey PE & Roth RA. Inflammatory Stress and Idiosyncratic Hepatotoxicity : Hints from Animal Models. Pharmacological Reviews. 2009; 61: 262-282.
- Di Meo S, Reed TT, Venditti P & Victor VM. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Medicine and Cellular Longevity. 2016; 44.
- Martin RM & Bachman MA. Colonization, infection, and the accessory genome of Klebsiella pneumoniae. Frontiers in Cellular and Infection Microbiology. 2018: 1-15.
- Alarcon-Aguilar FJ, Ellis PR, Bailey CJ, Eloff JN, Baynes KCR, Ghisalberti EL, et al. Traditional Medicines for Modern Times. In A. Soumyanath (Ed.), Traditional Medicines for Modern Times. 2006.
- Rai MK & Kon KV. Fighting Multidrug Resistance with Herbal Extracts, Essential Oils and Edited by (first). 2013.
- Selim KA, El-beih A, Abdel-rahman TM & El-diwany AI. Biology of Endophytic Fungi. Current Research in Environmental & Applied Mycology. 2012: 31-82.
- Strobel GA. Endophytes as sources of bioactive products. Microbes and Infection. 2003; 5: 535-544.
- Garcia A, Rhoden SA, Orlandelli RC, Azevedo JL & Pamphile JA. Antimicrobial Activity of Crude Extracts of Endophytic Fungi Isolated from Medicinal Plant Sapindus saponaria L. Journal of Applied Pharmaceutical Science. 2012; 2: 35-40.
- Kumar S, Aharwal RP, Shukla H, Rajak RC & Sandhu SS. Endophytic fungi : as a source of antimicrobials bioactive compounds. World Journal of Pharmacy and Pharmaceutical Sciences. 2014; 3: 1179-1197.
- Singh R, Singh D, Rathod V, Naaz SA, Kumar A, Siddique S, et al. Phytochemical Analysis and Antibacterial Studies of the Crude Extracts of Endophytic Fungi, Colletotrichum sp. and Alternaria sp. from the Medicinal Plant Tridax procumbens (L). International Journal of Natural Products Research. 2015; 5: 27-33.
- Adam SIY, Yahya AAN, Salih WMM & Abdelgadir WS. Antimicrobial Activity of the Masticatory Cola acuminat a Nut (Gooro). Current Research Journal of Biological Sciences. 2011; 3: 357-362.
- Mfouapon MH, Hzounda FBJ, Yimgang LV, Toghueo KRM, Fogue Soubgwi P, Eke P, et al. Isolation of endophytics fungi from Cola acuminata Schott & Endl , and antifungal activity against Candida Sp. European Journal of Biology and Biotechnology. 2020; 1.
- Salman A, Pomerantz A, Tsror L, Lapidot I, Moreh R, Mordechai S, et al. Utilizing FTIR-ATR spectroscopy for classification and relative spectral similarity evaluation of different Colletotrichum coccodes isolates. Analyst. 2012; 137: 3558-3564.
- Farmacia F & Farmaceutiche S. Innovative Immunoassay Formats For The Detection Of Food Contaminants And Pathogenic Bacteria. degli Studi di Bologna. 2006.
- Lecellier A. Caractérisation et identification des champignons filamenteux par spectroscopie vibrationnelle. Reims Champagne-Ardenne. 2013.
- Li Y, Kong D & Wu H. Analysis and evaluation of essential oil components of cinnamon barks using GC–MS and FTIR spectroscopy. Industrial Crops and Products. 2013; 41: 269-278.
- Stuart B. Biological Applications of Infrared Spectroscopy (D. J. ANDO, Ed.). sedney, Australia: John Willey and Sons. 1997.
- Stuart B. Infrared Spectroscopy: Fundamentals and Applications (John Wiley & Sons, Ed.). Analytical Tachniques in Science. 2004.
- Sun D. Principles of Infrared Spectroscopy. In D SUN (Ed.), Infrared spectroscopy for Food quality analysis and Control (1st ed.). Oxford, UK: Elsevier. 2009.
- Vrabl P, Schinagl CW, Artmann DJ, Heiss B & Burgstaller W. Fungal Growth in Batch Culture - What We Could Benefit If We Start Looking Closer. Frontiers in Microbiology. 2019; 10: 1-11.
- Cai MH, Zhou XS, Sun XQ, Tao KJ & Zhang YX. Statistical optimization of medium composition for aspergiolide A production by marine-derived fungus Aspergillus glaucus. Journal of Industrial Microbiology and Biotechnology. 2009; 36: 381-389.
- Chaichanan J, Wiyakrutta S, Pongtharangkul T, Isarangkul D & Meevootisom V. Optimization of zofimarin production by an endophytic fungus , Xylaria sp. Acra L38. Brazilian Journal of Microbiology. 2014; 293: 287-293.
- Damour H, Okoye FBC, Proksch P, Hakiki A, Mosaddak M, Hegazy MF, et al. Pretrichodermamide A and nafuredin from Trichoderma sp , an endophyte of Cola nitida. J. Mater. Environ. Sci. 2015; 6: 779-783.
- Jing X. Secondary Metabolites from the Endophytic Fungus Pestalotiopsis sp. JCM2A4 and its Microbe-Host Relationship with the Mangrove Plant Rhizophora mucronata. Düsseldorf. 2010.
- Strobel GA. Microbial gifts from rain forests. Can. J. Plant Pathol. 2002; 24: 14-20.