
Research Article
Austin J Pharmacol Ther. 2022; 10(1).1160.
Bioequivalence Estimation of Two Formulations of Cefixime Tablets among Healthy Chinese Volunteers under Fasting Condition
Hu Q1,2, Zhan W2, Ma X2, Huang L2 and Li X1*
1Department of Epidemiology & Biostatistics, and Center for Clinical Big Data and Statistics, Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, China
2Apeloa Pharmaceutical Co., Ltd., Hengdian, Dongyang, Zhejiang, China
*Corresponding author: Xiuyang Li, Department of Epidemiology & Biostatistics, and Center for Clinical Big Data and Statistics, Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China
Received: March 02, 2022; Accepted: March 25, 2022; Published: April 01, 2022
Abstract
Objective: To assess the bioequivalence of 200mg Cefixime tablets after oral administration to healthy adults under fasting condition.
Method: This study was an open-label, balanced, randomized singledose, two-treatment, two-sequence, two-period, crossover oral bioequivalence study in healthy adult, human subjects under fasting condition. Subjects were fasted overnight for at least 10.00 hours before scheduled time of start of dosing. Investigational Product, one tablet of the test formulation or one tablet of reference formulation (allocated as per the randomization schedule) was administered orally to each subject. The pharmacokinetic parameters maximum plasma concentration (Cmax), area under the plasma concentration-time curve (AUC0-t), AUC extrapolated to infinity (AUC0-8) was estimated to prove bioequivalence. Acceptance range for bioequivalence was 80.00%-125.00% for 90% confidence intervals of the geometric least square means ratio for Cmax, AUC0-t and AUC0-8.
Results: For the test formulation, the Cefixime Mean Cmax was 3891.31ng/ mL (vs. 3676.32ng/mL for reference), AUC0-t was 33299.52ng•h/mL (vs. 32182.07ng•h/mL) and AUC0-8 was 34234.88ng•h/mL (vs. 33162.95ng•h/mL). The 90% confidence intervals for the Geometric Least Squares Means ratios for Cefixime were 105.85% (90% CI: 98.73%-113.48%), 103.47% (90% CI: 96.34%-111.13%) and 103.23% (90% CI: 96.28%-110.69%) for Cmax, AUC0-t and AUC0-8 respectively, which are within the acceptance range of 80.00% to 125.00% for pharmacokinetic parameter Cmax, AUC0-t and AUC0-8 required for concluding bioequivalence between the test and reference formulations. There were no deaths or serious adverse events during the conduct of the study.
Conclusion: Test product when compared with the reference product meets the bioequivalence criteria in terms of rate and extent of absorption of cefixime after administration of single dose under fasting condition.
Keywords: Cefixime; Bioequivalence; Pharmacokinetics; Safety evaluation; Clinical trial
Introduction
Cefixime has a broad antibacterial spectrum and has antibacterial activity against some Gram-positive and negative bacteria, especially against Streptococcus (except Enterococcus), pneumococcus, and Gram-negative bacteria in Gram-positive bacteria. Neisseria gonorrhoeae, Branhamella, Escherichia coli, Klebsiella, Serratia, Proteus, Influenza, etc. have strong antibacterial effects, and their mechanism of action is bactericidal. It has strong stability to β-lactamase produced by various bacteria, and shows superior antibacterial power to β-lactamase producing bacteria. Clinically, this product is often used to treat acute attacks of chonic bronchitis caused by sensitive bacteria, acute bronchitis complicated by bacterial infection, bronchiectasis complicated infection, pneumonia; pyelonephitis, cystitis, gonococcal urethitis; acute biliary system Bacterial infections: cholecystitis, cholangitis; scarlet fever; otitis media, sinusitis [1].
When a normal adult takes 50, 100, and 900 mg (potency) once orally under fasting conditions, the serum concentration reaches the peak value (Cmax), which is 0.69, 1.13 and 1.95 μg/mL respectively after about 4 hours. The half-life of the blood concentration is 2.3 ~ 2.5 hours. When a single oral dose of 1.5, 3.0 and 6.0 mg/kg (potency) of this product in children with normal renal function, the blood drug concentration reaches the peak (Cmax) after about 3 to 4 hours, which are 1.14, 2.01, 3.97 μg/mL, respectively. The half-life of the drug concentration is 3.2 to 3.7 hours. Compare the pharmacokinetics of a single oral administration of 100mg cefixime in patients with moderate (30≦Ccr<60mL/min, n=3) and severe (10≦Ccr<30mL/min, n=4) renal failure patients study. After 6 hours of oral administration in patients with moderate renal failure, the plasma concentration reached the peak (2.04μg/mL), while in patients with severe renal failure; the plasma concentration reached the peak (2.27μg/mL) after oral administration for 8 hours. The blood drug concentrations were 0.71, 1.83 μg/mL, and the blood drug concentration half-lives were 4.15 hours and 11.05 hours, respectively. The half-life of patients with severe renal failure was longer. No antibacterial active metabolites were found in human serum and urine. Cefixime is mainly excreted though the kidneys. When healthy adults (fasting) take 50, 100, 200 mg (potency) of this product, the urinary excretion rate (0-12 hours) is about 20-25%, and the urinary drug concentration is 42.9 (4-6 hours), 62.2 μg/mL (4-6 hours), 82.7μg/mL (4-6 hours). In addition, in children with normal renal function, the urinary excretion rate of 1.5, 3.0, 6.0 mg/kg (potency) of this product is about 13-20% [1].
In this work, the bioequivalence between 2 formulations of the 200mg Cefixime tablets in healthy volunteers under fasting condition was conducted. The objective of this study is to compare the rate and extent of absorption of the generic (test) and branded (reference) formulations of 200mg Cefixime tablets after oral administration to healthy adults under fasting condition.
Subjects and Methods
This open label, balanced, randomized, single-dose, twotreatment, two-sequence, two-period, crossover oral bioequivalence study was conducted between March 2019 and April 2019.The study was conducted in compliance with protocol, Declaration of Helsinki and the moral, ethical and scientific principles stipulated by GCP. Ethical approval was obtained from Clinical Trial Ethics Committee of West China Second University Hospital, Sichuan University on 18 October 2018. 34 subjects were enrolled in the study.
Subjects
Prior to the start of the study, each subject was provided with a subject informed consent and procedures document that provided information on the investigational drug and the procedures and the potential risks of participating in the study by designated study personnel. Investigator discussed with volunteer about study related understanding and encouraged the volunteers for raising queries/ questions related to any aspect of the study and resolved the same, up to his satisfaction. After that, investigator reviewed the signed and dated consent document before enrolling the subject in study. All subjects underwent a screening procedure comprising medical history, clinical examination (Including vital signs (sitting blood pressure, oral temperature, radial pulse rate and respiratory rate), physical examination and systemic examination), recording of electrocardiogram and laboratory investigations of blood as well as urine not more than 14 days prior to first dosing. Subjects who were aged above 18 years and whose weight within clinically acceptable normal range according to normal values for Body Mass Index (19.0 to 26.0 kg/m² (both inclusive)) with minimum of 45kg and 50kg weight for female and male respectively were selected for study participation. Exclusion criteria were a history of allergy to cefixime or its preparation excipients, other cephalosporins and penicillins were excluded from the study or other situation assessed by investigators. Urine screen for drugs of abuse and alcohol breath test were performed on admission day of each study period for all subjects.
Subject design and treatments
This comparative bioavailability study, utilizing single dose of one test and one reference formulation was designed as crossover to minimize subject-by-subject variations. As bioavailability/bioequivalence studies do not typically require a double- blind study, this study design was open-label. As this was a bioequivalence study and there was no clinical efficacy assessment, a control group was not included. Furthermore, according to “Technical guidelines for research on human bioequivalence of chemical drug generic drugs with pharmacokinetic parameters as the endpoint evaluation index” [2], a randomized, open label, balanced, two-treatment, twoperiod, two-sequence, single dose, crossover, oral bioequivalence study design was considered the preferred design for comparative bioavailability studies.
Refer to previous relevant clinical trial literature reports [3,4], according to the coefficient of variation of 25%, the ratio of the test to the reference is 0.95, when a is 0.05, the power is 80%, and the equivalent confidence interval is set to 0.8-1.25, The sample size estimated using PASS11 software (Version 11.0.7) is 28 subjects, considering the dropout rate of about 15%, 34 subjects were included in the study.
All subjects required to fast (overnight) for at least 10.00 hours before scheduled time of dosing. Investigational Product, one tablet of the test formulation or one tablet of reference formulation (allocated as per the randomization schedule) was administered orally to each subject at scheduled dosing time and was instructed to swallow it with 240±2 mL of dosing water at ambient temperature in sitting posture. Subject was instructed not to chew or crush the Investigational Product but to consume it as a whole. Compliance for dosing was assessed by a thorough check of the oral cavity using torch and disposable spatula immediately after dosing. During each period subjects remained in the study center until blood samples had been taken 24h after dosing.
Pharmacokinetic evaluation
A total of 18 blood samples were collected during each period. The pre-dose blood sample of 4.0mL (0.00h) was collected within one hour before dosing and the post- dose blood samples of 5.0mL each was drawn at 0.5, 1.0, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 7.0, 8.0, 10, 12, 14, 24 hours following drug administration in each period [5]. After collection of blood samples from all the subjects at each time point, trained study- personnel centrifuged the samples at 2000g for 7 minutes at 4°C. After centrifugation, vacutainers were arranged as per subject number in rack/stand placed on wet ice bath. A validated UPLC/MS bioanalytical method developed for the quantification of Cefixime in Human Plasma was employed for subjects’ sample analysis. A calibration curve extending over the range from 62.5ng/mL to 5000ng/mL with a LLOQ of 62.5ng/mL was used in subject sample analysis of Cefixime. Plasma concentrations of cefixime were analyzed by LC-MS/MS method which was using multiple reaction monitoring (MRM) mode, and 13C3, 15N2-cefixime was used as the internal standard. After the plasma samples were precipitated with methanol, the supernatant was injected for analysis. The chromatographic column is Waters ACQUITY BEH C18 2.1×50mm, 1.7μm (mobile phase 0.1% formic acid aqueous solution and methanol). The limit of quantification was 62.5ng/mL.
A total of 1206 samples were analyzed during the study. Out of these, 134 samples were reanalyzed to establish incurred sample reproducibility (ISR). A total of 132 (98.5%) samples were within the acceptance criteria of ±20% differences between the mean of ISR sample value and original value.
Safety evaluation
Safety was assessed from the screening period to the end of the study. It was assessed through clinical examination, vital signs assessment, axillary body temperature, 12-lead electrocardiogram (ECG), chest X-ray (posterior-anterior view) recording, clinical laboratory parameters, (e.g. biochemistry, hematology, immunology and urine analysis), serum pregnancy test (for female subjects), subjective symptomatology and monitoring of adverse events.
Pharmacokinetic analyses
The pharmacokinetic parameters are derived individually for each analyzed subject from the plasma concentration vs. time profiles of Cefixime. The pharmacokinetic parameters Cmax, AUC0-t, AUC0-8 were considered the primary indicators of bioequivalence, whereas Tmax, t½, Kel, R² adjusted and AUC_%Extrap_obs were considered secondary parameters to demonstrate bioequivalence. Actual time points of the sample collection were used for the calculation of pharmacokinetic parameters.
Statistical analyses
Descriptive statistics were calculated and reported for the pharmacokinetic parameters of Cefixime. ANOVA, power and ratio analysis for ln-transformed pharmacokinetic parameters Cmax, AUC0-t and AUC0-8 are calculated and reported for Cefixime .Using two onesided tests for bioequivalence, 90% confidence intervals for the ratio of the geometric least-squares means between drug formulations are calculated and reported for ln-transformed pharmacokinetic parameters Cmax, AUC0-t and AUC0-8 for Cefixime.
Dataset for estimation of pharmacokinetic parameters Cmax, AUC0-t, AUC0-8, Tmax, t½, λz and AUC_%Extrap was planned to calculate using non-compartmental model by using Phoenix WinNonlin® 8.0. For Cefixime, the ln-transformed pharmacokinetic Cmax, AUC0-t and AUC0-8 were analyzed by analysis of variance (ANOVA) using PROC GLM in SAS® Software, Version 9.4.
Results
67 volunteers were screened, of whom 34 met the inclusion criteria. 1 subject was withdrawn and 33 subjects were completed both the periods of the study. Weight of the subjects was within clinically acceptable normal range according to normal values for Body Mass Index 19 to 26 kg/m² with minimum of 45kg weight. Age of the subjects was between 18 and 36 years (Table 1).
![]()
Characteristic
Mean±SD
Age (Years)
23.9±4.28
Height (cm)
165.58±8.865
Weight (kg)
60.29±8.562
BMI (kg/m2)
21.88±1.395
Table 1: Demographic data (n=34).
The mean plasma Cefixime concentration–time profiles were similar for the 2 formulations of Cefixime tablets. The Linear and semi-logarithmic plots of mean plasma concentrations versus time of Cefixime tablets are shown in Figure 1 and 2.
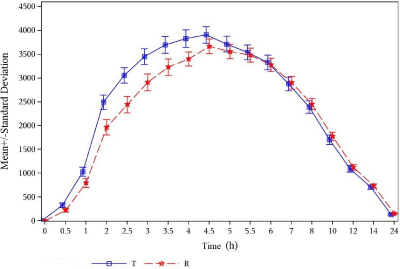
Figure 1: Linear plot of mean concentration vs. time for Cefixime.
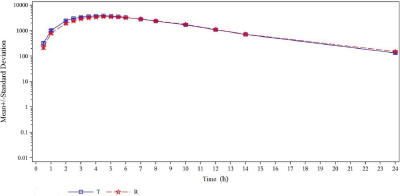
Figure 2: Semi-logarithmic plot of mean concentration vs. time for Cefixime.
The pharmacokinetic parameters Cmax, AUC0-t and AUC0-8 were considered the primary indicators of bioequivalence, whereas Tmax, λz, t½, and AUC_%Extrap were considered secondary parameters to demonstrate bioequivalence. The estimation of all the pharmacokinetic parameters was very close between the 2 formulations.
The adjusted geometric mean ratios (test/reference) of Cmax, AUC0-t and AUC0-8 for the 2 Cefixime formulations were within the allowed bioequivalence limit specified by the regulatory authorities (80%-125% for Cmax, AUC0-t and AUC0-8). The mean pharmacokinetic parameters estimated of reference and test tablets are shown in Table 2. The geometric least squares mean of Test Formulation (T) and Reference Formulation (R), its ratio (T/R) %, intra-subject variability, power and 90% confidence intervals for geometric least square mean ratio (T/R) obtained from the analysis of ln-transformed parameters Cmax, AUC0-t and AUC0-8 are shown in Table 3.
![]()
Pharmacokinetic Parameters(Units)
Mean ± SD (untransformed data)
Test product
Reference product
Cmax (ng/mL)
4004.61 ± 984.35
3772.29 ± 881.54
#Tmax (h)
4.50(2.5, 5.5)
4.50(3.5, 6)
AUC0-t (h*ng/mL)
34707.12 ± 10214.41
33468.44 ± 9083.48
AUC0-8 (h*ng/mL)
35661.22 ± 10459.49
34474.71 ± 9379.94
λz(1/h)
0.185 ± 0.04
0.175 ± 0.03
t½ (h)
3.854 ± 0.60
4.066 ± 0.62
AUC_%Extrap
2.723 ± 1.27
2.961 ± 1.27
#For Tmax median (min, max).
Table 2: Descriptive statistical results of pharmacokinetic parameters of cefixime tablets.
![]()
Parameters
Geometric least square means (n=33)
Intra-subject CV (%)
90% Confidence Intervals
Power (%)
Test (T)
Reference (R)
T/R (%)
LnCmax
3891.31
3676.32
105.85
16.8
(98.73,113.48)
99
LnAUC0-t
33299.52
32182.07
103.47
17.23
(96.34,111.13)
99.7
LnAUC0-8
34234.88
33162.95
103.23
16.82
(96.28,110.69)
99.8
Table 3: Results of bioequivalence evaluation of cefixime tablets.
Safety results
In general, the investigational products were well tolerated by healthy subjects. Four (04) adverse events (AEs) were reported by four (04) subjects during the conduct of the study. Adverse Events in this study were coded using MedDRA version 21.1. The Severity of all AEs were grade-I. The subjects were followed up until resolution of their AEs. The causality assessment was judged as possible for two (02) AEs, as unlikely for two (02) AEs. There were no deaths or serious adverse event during the conduct of the study.
Discussion
The objective of the study was to compare the rate and extent of absorption of Test Product (T) and Suprax® Cefixime tablets 200mgof Sanofi in healthy, adult, human subjects under fasting condition.
Data from total enrolled subjects (n=34) were used for pharmacokinetic analyses. One subject withdrew from study after dosing in period-I, hence data from 33 subjects who completed the study were used for equivalence statistical analyses. The design of the study was adequate to determine the pharmacokinetic parameters of the test and the reference formulations. A washout period of seven (07) days was kept between each consecutive dosing period to minimize the carry over effects and to ensure complete elimination of the drug from the body. In accordance with the regulation requirement, the study meets the bioequivalence criteria as 90% confidence interval for the ln-transformed pharmacokinetic parameter Cmax, AUC0-t and AUC0-8 is within the acceptance range of 80.00%-125.00%.
Conclusion
The Test Product (T) when compared with the Reference Product (R) (Suprax® Cefixime tablets 200mg of Sanofi) meets the bioequivalence criteria in terms of rate and extent of absorption after administration of single dose under fasting condition.
Declaration
Ethical approval: The study was approved by Clinical Trial Ethics Committee of West China Second University Hospital, Sichuan University.
Funding: This study received funding from Zhejiang Jutai Pharmaceutical Co. Ltd, which is the subsidiary of Apeloa Pharmaceutical Co., Ltd. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article. All authors declare no other competing interests.
Conflict of Interest: Qing Hu, Weiqiang Zhan, Xiaoying Ma, Liqing Huang are employed by Apeloa Pharmaceutical Co., Ltd, and they have equal contribution to manuscript revision, read, and approved the submitted version.
References
- Prescription instructions of cefixime capsules and granules, Choseido Pharmaceutical Co., Ltd. Japan.
- Technical guidelines for research on human bioequivalence of chemical drug generic drugs with pharmacokinetic parameters as the endpoint evaluation index, Circular 61 of 2016, Center for Drug Evaluation, NMPA. 2021.
- Center for drug evaluation and research, application number: 203195orig1s000, clinical pharmacology and biopharmaceutics review(s).
- Public assessment report of the Medicines Evaluation Board in the Netherlands, Cefixim Schluttig 200mg and 400mg, film-coated tablets Cefixim Schluttig 100mg/5ml, power for oral suspension Pharma Resources Dr. Schluttig GmbH, Germany.
- Parvin Zakeri-Milani, Hadi Valizadeh, Ziba Islambulchilar. Comparative Bioavailability Study of Two Cefixime Formulations Administered Orally in Healthy Male Volunteers. Arzneimittelforschung. 2008: 58.