
Research Article
Austin J Pharmacol Ther. 2023; 11(2): 1176.
“Simvastatin’s Mitochondrial Defenses against Angiotensin II-Induced Heart Failure”
Hari Prasad Sonwani*
Apollo College of Pharmacy, Anjora, Durg (C.G), India
*Corresponding author: Hari Prasad Sonwani Apollo College of Pharmacy, Anjora, Durg (C.G), India. Email: harisonwani10@gmail.com
Received: October 03, 2023 Accepted: November 09, 2023 Published: November 16, 2023
Abstract
Background and Aims: Heart failure is one cardiovascular condition that might progress due to mitochondrial dysfunction. In chronic heart failure, 3hydroxy-3-methylglutaryl CoA reductase inhibitors (statins), which prevent the production of ROS, have cardioprotective benefits. However, it is still unknown how statins can protect the mitochondria in heart failure Experimental Strategy: Angiotensin II (1.5 mg/kg/day) or co-administered simvastatin (oral, 10 mg/kg/day) were given to rats for 14 days, after which the treatment was withdrawn. Wheat germ agglutinin staining and echocardiography were used to analyze the structure and function of the heart. Transmission electron microscopy was used to analyze the shape of the mitochondria as well as the numbers of lipid droplets, lysosomes, autophagosomes, and mitophagosomes. After stimulating human cardiomyocytes, flow cytometry was used to assess changes in intracellular ROS and mitochondrial membrane potential (m). and, respectively, JC1 staining. By using immunohistochemistry and western blotting, apoptotic proteins that are associated to autophagy, mitophagy, and mitochondrial regulation were identified. Key outcomes Simvastatin mitigated the disruption of m and dramatically decreased ROS generation. Simvastatin stimulated autophagy and mitophagy, caused lipid droplets to accumulate, and provided energy for maintaining mitochondrial function and impeded apoptosis that was mediated by mitochondria. According to these results, simvastatin-mediated mitochondrial protection prevents heart failure by modifying antioxidant status and enhancing energy sources for autophagy and mitophagy, which prevent mitochondrial damage and cardiomyocyte apoptosis. Final Thoughts and Implications Mitochondria are crucial in controlling the course of heart failure. Simvastatin reduced angiotensin II-induced heart failure through mitochondrial preservation and may offer a new treatment for heart failure prevention.
Keywords: Cardiovascular; Simvastatin; Echocardiography; Cardiomyocytes
Introduction
Heart Failure (HF) is a condition that affects the entire world and is brought on by the aging global population. According to Bhatt and Butler (2018), HF is a frequent reason for individuals over 65 to be admitted to the hospital and will have a significant financial impact on healthcare globally. The production of ROS, mitochondrial dysfunction, and reduced cardiac contractility are typical pathological findings in HF [40]. One of the frequent reasons why HF progresses is pressure overload caused by angiotensin II (Ang II). According to studies, mito-chondrial dysfunction is a major factor in the development of HF [59]. However, there are currently no clinically proven ways to stop or even stop the progression of HF. The renin-angiotensin system in the body generates the peptide Ang II. Regulates ROS generation, mitochondrial dysfunction, pro-inflammatory cytokine expression, autophagy, apoptosis, and pathophysiology of the cardiovascular system, including hypertension and Heart Failure (HF) [51]. There are a lot of mitochondria in heart cells. Cardiomyocytes employ efficient preventative strategies to preserve mitochondrial homeostasis through mitochondrial quality management, which involves the regulation of mitochondrial dynamics and mitochondrial autophagy (also known as mitophagy) [16]. Recently, cardiovascular illnesses such cardiomyopathy and HF have been linked to mitochondrial quality, morphology, and function [25]. Mitophagy is the process by which damaged mitochondria are broken down. During this process, damaged mitochondria are consumed by autophagosomes and then broken down in lysosomes [50]. Additionally, it was recently shown that Ang II causes cardiomyocytes to engage in autophagy, with possible effects on HF linked to Ang II [28]. The stability of cardiomyocyte ATP production affects the progression of cardiac illness and mito chondrial respiratory function in addition to maintaining the cellular energy state [47]. One of the primary energy sources in cardiomyocytes is lipid. Cardiomyocytes increase their fatty acid content when heart injury is caused by hypoxia, ischaemia, or stress overload. According to Minami et al. (2017), acid (FA) intake results in intracellular Lipid Droplets (LDs), which are then hydrolyzed for usage by lysosomes or peroxisomes. Recent research has shown that the lysosome route is essential for the conversion of lipids to FAs [1,41]. Additionally, Dupont et al. (2014) showed that LD accumulation could encourage autophagy because LDs provide lipid precursors for the developing autophagosome membrane. According to Lee, Zhang, Choi, and Kim (2013), LD formation is a global stress response that is triggered by cardiac mitochondrial dys-function. In addition, Yokoyama et al. (2007) showed that taking a statin caused the cytosolic LDs to build up, giving the cell extra energy. Inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase, also known as statins, are a common class of medications used to treat Overproduction of cholesterol [38]. In addition to their effects on lipid levels, statins have been shown to have anti-inflammatory, anti-oxidative, and anti-cancer properties [28,52]; they also play a role in endothelial regulation and have an impact on cell autophagy and apoptosis [42,46]. However, there are still many issues that need to be researched further in relation to simvastatin's function in the heart. To determine whether simvastatin's control of We used an HF animal model and cultured Human Cardiomyocytes (HCMs) to investigate the preventive effects of simvastatin in Ang II-induced mitochondrial damage. Mitophagy participates in mitochondrial protection in HF. By controlling LD accumulation, lysosomal activation to destroy LDs, inhibiting ROS production, and preventing alteration of mitochondrial membrane potential (m), simvastatin reduces Ang II-mediated HF, according to our findings. As a result, there is an increase in mitophagy, which prevents the activation of apoptosis by the mitochondria.
Methods
Treatment of Animals
The animal treatments were carried out in a facility that was approved by the Association for Assessment and Accreditation of Laboratory Animal Care International and strictly adhered to Taiwanese law. Eight-week-old male Sprague-Dawley rats were kept in a specified pathogen-free environment with a 12/12-hour light/dark cycle.having free access to regular food and water, as well as a temperature-controlled room. Throughout the trial, all rats were weighed once a week, and three experimental groups (each with six rats) were randomly selected. After isoflurane anaes- thesia (1.5%), each group received a subcutaneously implanted Alzet® osmotic pump (Durect Corporation, Cupertino, CA, USA; infusion rate of 0.5 μl·hr-1) with (a) PBS; control group, (b) Ang II (1.5 mg·kg-1·day-1; , Ang II group or (c) Ang II infusion and co-administration of simvastatin (oral, 10 mg·kg-1·day-1, Merck), AngII+SIM group. After being administered for 14 days, the infusions were ceased for the next 14. The rats were put to death (with CO2) on day 28, and the hearts were immediately taken. Upon completion of the experiment weight of the entire heart, the coronal heart portion (from apex to apex), the lung, and the gastrocnemius muscle were also recorded throughout this time. The authors claim that every attempt was made to reduce both the quantity and severity of the suffering experienced by the animals.
Echocardiography
Animals were given isoflurane (Panion & BF Biotech Inc., Taiwan) anesthesia before having M-mode transthoracic echocardiography performed utilizing an iE33TM imaging equipment and an S124 (12-4 MHz) pediatric probe. large sampling frequency (150 mm/s). At the level of the left atrium, two-dimensional targeted M-mode echocardiographic pictures were taken. to measure the left ventricular mass (LVM), LVM contractility, left ventricular ejection fraction (EF), fractional shortening (FS), left ventricular internal diameter in diastole, and left ventricular internal diameter in systole. For each assessment, three cycles were measured, and the average values were computed. The formula LVM (g) = 0.8 [1.04 (IVS + PP + LVEDD)3 LVEDD3] + 0.6 was used to calculate LVM (g), while Teichholz's formula was used to determine EF.
Oil Red O staining, germ agglutinin staining, immunohistochemistry, TUNEL labeling, and histology
As previously described [48], portions of the heart were fixed with 4% paraformaldehyde, wax embedded, and sectioned (5 m thick) for H&E staining (Invitrogen, Carlsbad, CA, USA). The size of cardiomyocytes, and Wheat germ agglutinin (WGA) staining and ImageJ analysis (RRID: SCR_003070, NIH, Bethesda, Rockville, MD, USA) were used to estimate length. Oil Red O staining was quantified to determine the lipid content (Bio Vision, Mountain View, CA, USA; Catalog #K58024). The samples were stained with haematoxylin, washed with distilled water, and then were washed three more times with 60% isopropanol for five minutes each while being gently rocked. They were then replaced in 24 well plates for measurement after being removed by Oil Red O stain with 100% isopropanol for 5 min with gentle shaking. After subtracting the background signal using 100% isopropanol as a background control, the absorbance was measured at 495 nm. The methods in this investigation that were based on antibodies abide by the British Journal of Pharmacology's recommendations. Measurements of autophagic or apoptotic protein expression were made using immunohistochemical staining and western blotting. Tissue sections were first incubated in blocking buffer (0.5% BSA, 0.05% Tween 20, and PBS) for 1 hour at room temperature, then specific primary antibodies against Bcl2 (1:100, sc7382, RRID: AB_626736, Santa Cruz Biotechnology, Santa Cruz, CA, USA), cytochrome c The substance According to manufacturer's protocol, staining was created using a 3,3'-diaminobenzidine detection system (Catalog #760124, Ventana Medical Systems, Tucson, AZ, USA) and counterstained with hematoxylin. According to the manufacturer's instructions, TUNEL assays were carried out using a TUNEL assay kit (In Situ Cell Death Detection Kit, Catalog 11684795910, Roche, Mannheim, Germany). A fluorescent microscope (Leica, Wetzlar, Germany) was used to view the cell nuclei after they had been counterstained with DAPI, cleaned, mounted with VECTASHIELD® mounting media (Vector Laboratories, Burlingame, CA, USA), and washed.
Electron Microscope that Uses Field-Emission Tomography
According to previous descriptions [45], field-emission trans mission electron microscopy (FETEM) was implemented. Briefly stated, tissue samples were preserved for two hours at 4°C with 2.5% glutaraldehyde. Following a thorough cleaning, the samples were post-fixed for two hours in 1% osmium tetroxide, dehydrated in graded acetone, infiltrated, and finally embedded in epoxy resin. Using a Leica, ultrathin 70 nm slices were cut. At an accelerating voltage of 80 kV, samples were cut with a microtome (Leica RM2165, Japan) and analyzed using a FE-TEM (HITACHI HT-7700, Japan).
Measuring the level of Mitochondrial Damage
A mitochondrion is made up of five different components and has a double-membrane structure made of proteins and phospholipid bilayers: the mitochondrial outer membrane, (b the inner mitochondrial membrane, (c) the cristae space (produced by infoldings of the inner membrane), (d) the cristae space (the space within the inner membrane), and (e) the matrix (the space within the inner membrane). The shape of the mitochondrial cristae directly reflects the health of the mitochondria, and FE-TEM images demonstrate various degrees of mitochondrial damage: Score 1: cristae are joined to the intermembrane space by well-defined, many crista junctions and cristae, indicating healthy mitochondria (well-defined, undamaged, ordered membranes); Score 2: early stages of enlarged mitochondria (sometimes enlarged cristae, rather erratic); Megamitochondria receive a 3 (severe deformities, significant swelling and disarray of the cristae, and discontinuous membrane and cristae); Score 4: incredibly large, enlarged matrix mitochondria (membranes and cristae separated into particles to Score 5: vacuolization (delamination of the inner and outer mitochondrial membranes, absence of cristae, vacuolization); and Score 4: widespread mitochondrial ghosts.
Culture of Cells
The cells were grown in accordance with earlier instructions [45]. HCMs were cultured in cardiac myocyte medium (ScienCell Research Laboratories; Catalog #6200; Carlsbad, CA, USA) with 5% FBS (Life Technologies; Ref. 10437-028; Lot 1700200), 1% cardiac myocyte growth supplement (ScienCell Research Laboratories), and 1% penicillin/streptomycin solution (Life Technologies; Ref. 15140-122; Lot 1881449) added. The culture media was changed every 4 to 5 days while the cells were incubated at 37°C in a 5% CO2 environment. The cells were utilized exclusively between Pas- sages 3 and 9.
Measurement in Millimeters
A JC-1 m detection kit (ThermoFisher Scientific, Waltham, MA, USA; Catalog M31152) was used to assess the Ang II-induced changes in m. in the past [4]. HCMs (1 103) were seeded on the cover slip for 48 hours, after which they were exposed to 2 gml of JC1 at 37°C for 20 min. They were subsequently treated with Ang II or Ang II + simvastatin for 2 hours. A BD LSR II flow cytometer (BD Bioscience, Singapore) or an Olympus FV1000 confocal microscope (Olympus, Tokyo, Japan) was used to analyze and take pictures of the cells. A rough estimate of JC1's excitation peak is 488 nm. For monomeric and J-aggregate forms, the estimated emission peaks are 529 and 590 nm, respectively.
ROS Measurement
Simvastatin (0.5 M, Sigma Aldrich, St. Louis, MO, USA) was pretreated with or without the addition of mitoSOXTM (5 M, ThermoFisher Scientific) for 2 hours in HCMs. ThermoFisher Scientific; Catalog 6827) for 30 min at 37°C and then treated with Ang II (10 M) for 1.5 hrs. 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA; 20 M, ThermoFisher Scientific; Catalog 6827) for 10 min at 37°C. Using flow cytometry, the mitochondrial and intracellular ROS levels were assessed. The fluorescence emission and excitation at 510 and 580 nm (MitoSOXTM) and 490 and 520 nm (H2DCFDA), respectively, were measured using the BD LSR II instrument (BD Bioscience).
Western Blot Examination
Western blot analyses were carried out as previously explained (Kuo et al., 2016). The Lowry assay was used to assess the protein levels in cell or tissue lysates. then 30g of protein samples
Depending on the molecular weight of the proteins of interest, they were separated using 7.5%, 10%, or 12.5% SDS-PAGE, and then electroblotted onto a nitrocellulose membrane. The membranes were then incubated with specific primary antibodies against Bcl2 (1:500, sc-7382, Santa Cruz Biotechnology), cytochrome c (1:1,000, ADI-AAM-175, Enzo Biochem), caspase 3 (1:1,000, #9662, Cell Signaling Technology), and LC3I/II (1:500, sc-7382, Santa Cruz Biotechnology).
Parkin, p62 (1:500, GTX100685, Gene Tex), GTX127375, Gene Tex GAPDH (1:2,000, sc137179, Santa Cruz Biotechnology), PINK1 (1:1,000, sc32282, Novus Biotechnology), and GAPDH were diluted before being detected using secondary antibodies that were HRP-conjugated. Fluorography and an improved detection kit (ECL, GE Healthcare Life Sciences, Buckinghamshire Amersham Pharmacia International) were used to visualize the signals.
Information and Analysis
The data and statistical analysis adhere to the British Journal of Pharmacology's guidelines for experimental design and pharmacology analysis. The results were analyzed using ANOVA, followed by Dunnett's post hoc tests, and are shown as the means SEM of each group. SigmaStat version 3.5 (RRID: SCR_010285, Systat Software Inc., Chicago, IL, USA) was used to generate all statistics, and P .05 was regarded as statistically significant.
Target and Ligand Nomenclature
Key protein targets and ligands are permanently archived in the Concise Guide to Pharmacology 2017/18 (Alexander, Cidlowski et al., 2017; Alexander, Fabbro, 2018) and are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to Pharmacology (Harding et al., 2018).
Results
LVM contractility (Figure 2d) and (Figure 2c). The left ventricular internal diameter, on the other hand, demonstrated that Ang II did not directly alter cardiac dilation. Simvastatin enhances cardiac performance in Ang II-induced heart failure The body weight of the study's experimental animals was measured at both the beginning (8 weeks) and the end (12 weeks), and it was determined to be 358.5 10.9 g 434.8 34.4 g in the control group, 354.6 11.8 g 399.6 13.7 g in the Ang II group, and 354.6 11.8 g 399.6 13.7 g in the Ang II group. In the Ang II + SIM group, the weights were 355.5 13.4 g 422.4 33.6 g. The most prevalent anatomical abnormality known as cardiac cachexia is severe HF, left ventricular hypertrophy, and skeletal muscle wasting (Delafontaine & Akao, 2004). Figure 1 displays the data that were gathered after the experiment. Body weight and gastrocnemius muscle weight are presented in Figures 1a,b, whereas Figure 1c depicts whole-heart imaging and H&E staining to identify histological changes. WGA staining and quantification were used to demonstrate that the treatment with Ang II did enhance cardiomyocyte size when compared to the control or simvastatin-treated groups (Figure 1d). The preventive benefits of simvastatin were subsequently investigated using echocardiography, which clarified the structure and function of the heart. Simvastatin and Ang II were administered to rats for 14 days, followed by 14 days of recovery. The rats' EF and FS were lowered at day 28 (Figure 2a and Figure 2b). LVM (Figure 2 eter in diastole (Figure 2e) and left ventricular internal diameter in systole (Figure 2f) values also rose (Figure 2b). In the last stages of HF, significant consequences such cardiac hypertrophy and pulmonary oedema frequently manifest [7,33]. Rat heart/body weight ratios (Figure 2g), lung/body weight ratios (Figure 2h), and gastrocnemius muscle weight/body weight ratio (Figure 2i) were calculated at the conclusion of the experiment, as in previous studies [22,25], in order to track the progression of Ang II-induced HF. Our findings demonstrated that Ang II significantly increased pulmonary oedema and cachexia in rats, which weren't seen in the SIM group plus Ang II. According to these findings, simvastatin supplementation reduced Ang II-induced ventricular hypertrophy, EF/FS decline, and pulmonary oedema. Simvastatin prevents in vivo myocardial mitochondrial damage caused by Ang II.
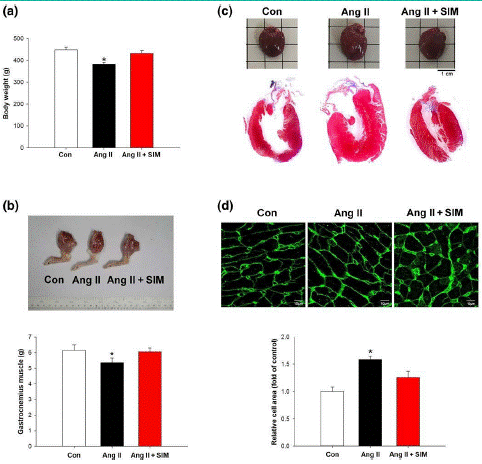
Figure 1: Simvastatin attenuates Ang II-induced cardiac hypertrophy in vivo. Male Sprague–Dawley rats were treated with Angiotensin II (Ang II, 1.5 mg·kg-1·day-1) or Ang II + simvastatin (SIM, oral, 10 mg·kg-1) for 28 days. Cardiac cachexia was determined by (a) body weights (n = 6 per group) and (b) gastrocnemius muscle weight (n = 6 per group). Left ventricle hypertrophy was determined by (c) H&E staining (n = 6 per group) and (d) WGA staining (n = 6 per group). (d) Relative folds were determined by comparing with the control (Con) group. *P < .05, significantly different from Con.
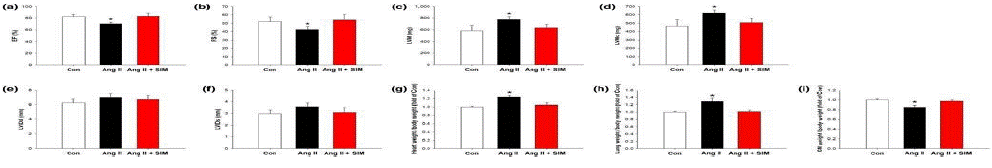
Figure 2: Simvastatin suppresses Ang II-induced heart failure in vivo. Cardiac function was examined by M-mode echocardiography: (a) ejection fraction (EF; n = 6 per group), (b) fractional shortening (FS; n = 6 per group), (c) left ventricular mass (LVM; n = 6 per group), (d) left ventricular mass contractility (LVMc; n = 6 per group), (e) left ventricular internal diameter in diastole (LVIDd; n = 6 per group), (f) left ventricular inte drnal diameter in systole (LVIDs; n = 6 per group), (g) heart weight normalized to body weight (n = 6 per group), (h) lung weight normalized to body weight (n = 6 per group), and (i) gastrocnemius muscle (GM) weight normalized to body weight (n = 6 per group). *P<.05, significantly different from control (Con).
According to Coughlin, Morrison, Horner, and Inman (2015), the structure and health of the mitochondria are directly reflected in the mitochondrion. Our FETEM results revealed a significant change in the morphological
After exposure to Ang II, mitochondria emerge and a matching histology score increases, both of which indicate mitochondrial damage (Figure 3a). In Section 2, the technique for measuring mitochondrial damage is explained.
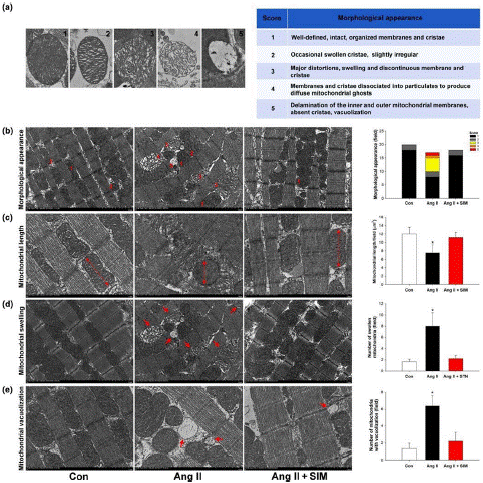
Figure 3: Simvastatin suppresses Ang II-induced cardiac mitochondrial damage in vivo. (a) Scoring criteria and examples of Ang II- damaged mitochondria (n = 6 per group). To determine whether simvastatin has mitochondrial protective effects in Ang II-damaged mitochondria, the (b) morphological appearance (n = 6 per group), (c) mitochondrial length (n = 6 per group), (d) number of swollen mitochondria (n = 6 per group), and (e) number of mitochondria with vacuolization (n = 6 per group) were measured by FE-TEM analysis. *P < .05, significantly different from control (Con).
Simvastatin was examined using FE-TEM analysis and mitochondrial score in the cardiac tissue of the control, Ang II, and Ang II + SIM groups to determine if it has mitochondrial protective effects in Ang II-mediated HF. At Day 28 (the end of the experiment), the appearance of the mitochondria was examined by FE-TEM (Figure 3b), and we then calculated the lengths of the mitochondria, the number of inflated mitochondria, and the number of mitochondria with vacuolization. The findings demonstrated that Ang II caused the development of large megamitochondria (Score 3), shortening of mitochondrial lengths, increasing of mitochondrial swelling, and compared to the control or Ang II+SIM groups, there was an accumulation of vacuolized mitochondria.
Simvastatin's effects on Ang II-mediated ROS generation, m depletion, and mitochondrial-mediated apoptosis are discussed in section 3.3.
Damaged mitochondria have been linked to a number of cardiovascular illnesses, and mitochondria are a key location of ROS formation [32]. Figure 4a and 4b demonstrate that HCMs treated to Ang II (10 M for 24 hr) had considerably higher amounts of super-oxide and total ROS in their mitochondria. These measurements were done using flow cytometry and MitoSOX fluorescence. The data come from three different investigations. As opposed to this, pretreatment with simvastatin (0.5 M for 2 hr) decreased mitochondrial superoxide to levels comparable to the untreated controls and considerably decreased total ROS content. In HCMs that were just exposed to Ang II. A typical cause of is excessive intracellular or mitochondrial ROS generation. Diminished m. In this investigation, cationic dye JC1 was used to stain HCMs, and m was measured using confocal microscopy (Figure 4c). The mitochondrial m was indicated by the JC1 signal (red color, aggregates, high potential; green color, monomers, low potential). Simvastatin pre-treatment successfully mitigated this Ang II-induced decrease in m, as seen by peak JC-1 fluorescence intensities (red, 590 nm; green, 530 nm). Ang II alone generated a significant reduction in m.
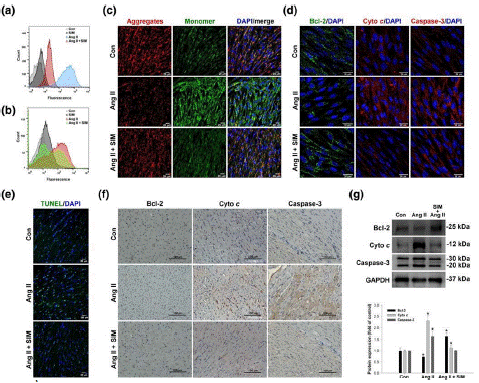
Figure 4: Simvastatin inhibits Ang II-induced ROS, δψm disruption, and mitochondrial-mediated apoptosis. Cultured HCMs were treated with Ang II (10 μM for 1.5 hr) or Ang II + simvastatin (pretreatment, 0.5 μM for 2 hr), and then the (a) mitochondrial superoxide (MitoSOX Red; n = 5) and (b) intracellular ROS (DCFH-DA) production were determined by flow cytometry (n = 5). Data are representative of three independent experiments. Cultured HCMs were treated with Ang II (10 μM for 2 hr) or Ang II + simvastatin (pretreatment, 0.5 μM for 2 hr), and then the HCM δψm was determined by measuring changes in JC-1-derived fluorescence from red (high potential, J-aggregrates) to green (low potential, monomeric) using confocal microscopy. Data are representative of three independent experiments, and (c) the scale bar in each image is 20 μm (n = 5). Cultured HCMs were treated with Ang II (10 μM for 24 hr) or Ang II + simvastatin (pretreatment, 0.5 μM for 24 hr), and then the (d) mitochondrial outer membrane protein: Bcl-2 (green; nucleus, blue), mitochondrial intermembrane/intercristae spaces protein: cytochrome c (cyto c; cleavage from, red; nucleus, blue), and pro-apoptotic protein caspase-3 (red; nucleus, blue) expression were measured by confocal microscopy (n = 5). In vivo, simvastatin attenuation of Ang II-induced apoptosis was confirmed by (e) TUNEL staining (green; nucleus, blue; n = 6 per group), (f) immunohistochemistry, and (g) western blot analysis (n = 6 per group). (g) Relative folds were determined by comparing with the control (Con) group. Qualitative data shown are representative of three independent experiments. *P < .05, significantly different from Con.
The effects of Ang II were first evaluated in cultured HCMs by measuring expression of the apoptosis-associated proteins, Bcl2, cytochrome c, and caspase 3 by immunofluorescence labelling (Figure 4d). This allowed researchers to identify whether mitochondrial damage-associated apoptosis was present. We used TUNEL labeling to confirm in vivo that Ang II caused cardiomyocyte apoptosis. Western blot analysis (Figure 4g) and immunohistochemistry staining (Figure 4f). According to our data, Ang II caused apparent DNA fragmentation when compared to the Ang II + SIM and control groups. Simvastatin is an additional Immunohistochemistry and western blotting revealed decreased Ang II-mediated cytochrome c release and caspase-3 activation.
Simvastatin controls the expression of lysosomes and LDs in cardiac tissue [3,4]. FAs are stored in LDs as triacylglycerol by cells. In order to increase the amount of energy available to the cell, adaptive LD production and FA metabolism in the mitochondria might be triggered by stress or nutritional deprivation [46]. Oil Red O staining was used to measure LD formation in order to examine the impact of simvastatin on mitochondrial energy supply (Figure 5a). This result was quantified using FE-TEM (Figure 5b), and FE-TEM was also utilized to examine the performance and distribution of lysosomes (Figure 5c). In rat cardiomyocytes, our findings revealed a few tiny LDs under basal conditions, whereas Ang II enhanced the number of LDs located close to the mitochondria. Furthermore, Comparative to the control and Ang II alone groups, adding simvastatin dramatically increased the amount of LDs positioned between mitochondria. Simvastatin addition also markedly raised the amount of lipophagy and the number of lysosomes in comparison to the control and Ang II alone groups. These findings collectively show that simvastatin treatment causes an accumulation of cytosolic LDs, which gives the cell additional energy to fend off harm from Ang II.

Figure 5: Simvastatin promotes mitophagy against Ang II-induced mitochondrial damage in vivo. To explore mechanisms of protection by simvastatin, (a) autophagosomes (red arrows) and (c) mitophagosomes (red arrows) were identified and quantified by FE- TEM analysis (n = 6 per group). Representative images are shown in (a) and (c), and quantitative plots are shown in (b) and (d); n = 6 per group. *P<.05 versus control (Con). Expressions of mitophagy-associated proteins LC3-I/II, p62, PINK1, and Parkin were confirmed by (e) immunohistochemistry (n = 6 per group) and (f) western blot analysis (n = 6 per group). *P<.05, significantly different from control.
Simvastatin encourages mitophagy to protect mitochondria from Ang II-induced damage in vivo [3,5]
Simvastatin-mediated mitophagosome/autophagosome production might have helped cells exposed to Ang II keep their mitochondria. To rule out this hypothesis, we used FE-TEM to analyze and determine the existence of autophagosomes and mitophagosomes in cardiac tissue (Figure 6c,d). Analysis. In contrast to the control group, treatment with Ang II alone or Ang II + SIM increased the number of autophagosomes, as seen in Figure 6a,b. However, compared to the control and Ang II only groups, the number of mitophagosomes was significantly higher in the Ang II + SIM group. The expressions of the mitophagy-associated proteins LC3-I/II, p62, PINK1, and Parkin were examined by immunocytochemistry labeling (Figure 6e) and western blotting (Figure 6f) to ascertain if simvastatin regulates mitophagy to protect mitochondria. According to our results, Ang II therapy alone or in combination with SIM enhanced LC3-I/II expression, but only the combination of Ang II + SIM significantly raised p62, PINK1, and Parkin protein expression when compared to that of the other treatments groups. Overall, our results imply that simvastatin controls mitophagy to prevent the progression of Ang II damage to the mitochondria in the myocardium.
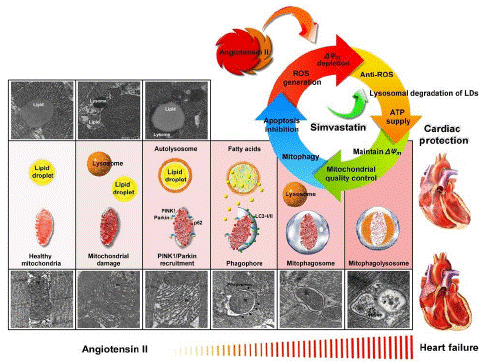
Figure 6: Summary scheme of the mitochondrial protection mechanism of simvastatin in Ang II-induced HF. Simvastatin could reduce ROS generation, regulate LDs and lysosome levels to provide energy to maintain Δψm, regulate mitochondrial quality control to promote mitophagy, and prevent mitochondrial-regulated apoptosis.
Conclusion and Discussion
In this investigation, we first established that simvastatin inhibited Ang II-induced HF by lowering mitochondrial and intracellular ROS production, increasing the amount of LD and lysosomes to supply energy for maintaining m, and promoting mitophagy and preventing mitochondria-mediated apoptosis.
In line with earlier observations, Ang II alone had a direct impact on cardiac tissues, causing cardiomyocyte hypertrophy and dysfunction [35]. A low dose of Ang II (1.1 mg kg 1 day 1), as stated by Dai et al. (2011) and discovered by echocardiography at Day 28 after daily administration, was used in our exploratory experiments. Administration, that HF was not caused by this dose (results not presented). As a result, we increased the dosage of Ang II to 1.5 mg/kg/day [47], gave it for 14 days, and then stopped treating the animals for the next 14 days. At Day 28, using this protocol, we discovered ultrasound evidence of HF. As a result, this dose was used in the current investigation.
According to Bjorkhem Bergman, Lindh, and Bergman (2011), the typical concentration of statins in human serum is only 1–15 nM.
Only 0.01–0.5 nM of the free fraction of statin is pharmacologically active. The majority of the information that is now available to support the claimed pleiotropic effects of statins is derived from in vitro experiments where significantly greater statin concentrations (1-50 M) were employed. Based on these findings, we conducted our in vitro tests with a concentration of 0.5 M simvastatin. We were able to demonstrate through our in vivo tests that chronic (14-day) Ang II administration caused left ventricular hypertrophy (as determined by WGA and H&E staining) as well as a decline in EF and FS. By constantly administering Ang II (1.5 mg kg 1 day) for 14 days using osmotic micro pumps, Aubdool et al. (2017) found that mice might develop hypertension, cardiac hypertrophy, and hypertensive HF. During this investigation, we didn't measure the alterations in animal BP as a result of our attention being on HF with hypertension. Choi et al. (2017) showed that cardiac fibrosis could be generated by administering oral Ang II (1 mg kg 1 day) for 15 days. In our investigation, we adjusted their procedures and were able to successfully induce HF and myocardial fibrosis in rats. According to our findings, simvastatin prevented Ang II-induced cardiac perivascular and interstitial fibrosis (Figure S1). However, it should be noted that, after computation, the dose utilized in vivo in our investigation was greater than the dose typically used in humans [17]. Monitoring potential liver and muscle toxicity brought on by simvastatin treatment would be necessary for the therapeutic application of our findings.
Ang II-triggered Arteriolar constriction, an increase in extracellular fluid volume, and pulmonary edema were all effects of HF. Muscle loss and weight loss are common signs of cardiac cachexia, which frequently occurs as HF progresses [33]. Simvastatin significantly reduced the EF and FS declines, pulmonary oedema, and cardiac cachexia caused by Ang II. Simvastatin reduces the development of hypertension, cardiovascular hypertrophy, increased heart weight index, and increased carotid cross-sectional area by Ang II by reduction of ROS formation, as demonstrated by Delbosc, Cristol, Descomps, Mimran, and Jover (2002). Simvastatin has recently been discovered to prevent cardiac hypertrophy via activating PPAR-dependent pathways [54], inhibiting RhoA/Ras-ERK pathways [3], or modulating the JAK/STAT pathway [55]. According to Zablocki and Sadoshima (2013), Ang II increases the levels of systemic ROS and mitochondrial oxidative stress, both of which have been linked to the pathophysiology of HF. According to flow cytometry and JC-1 labeling, exposure to Ang II led to the formation of cellular and mitochondrial ROS as well as a decrease in m in cardiomyocytes. Ang II decreases m, causing the opening of mitochondrial permeability transition pores, mitochondrial swelling, and activation of mitochondrial-mediated apoptosis [9,53]. Increased permeability, vacuolization, and the formation of intra-mitochondrial amorphous densities are not reversible after Ang II injection, while myocardial mitochondrial swelling is (Rosca & Hoppel, 2013). According to our findings, simvastatin reduced the significant swelling and vacuolization of the mitochondria caused by Ang II. Simvastatin also preserved mitochondrial length and dynamics by controlling mitochondrial quality (Ueta, Gomes, Ribeiro, Cardiomyocytes switch to mito-chondrial FA-driven oxidative phosphorylation in response to stress or nutritional deprivation to produce ATP. To do this, FAs, including those kept in LDs, must be transferred into the mitochondria [37]. Simvastatin markedly enhanced the amount of LDs found in close proximity to mitochondria. To maintain contractile and cardiac functioning, cardiac mitochondria create ATP [43]. According to a recent study, impaired FA oxidation or uncontrolled myocardial absorption of fat caused cardiac lipotoxicity in people with Type 2 diabetes and obesity [39].
Cardiomyocytes control mito- chondrial dynamics, biogenesis, and mitophagy to prevent mitochondrial damage [27]. Mitophagy, the process by which damaged or malfunctioning mitochondria are eliminated, increases cell survival under restricted environments by removing harmful protein aggregates and damaged organelles. Boundaries [40]. According to Zhao et al. (2014), Ang II causes apoptosis and mitophagy via increasing the generation of ROS. In this study, compared to the control and Ang II groups, simvastatin and Ang II increased the expression of the mitophagy proteins LC3-I/II, p62, Parkin, and PINK1. Simvastatin also reduced the activation of caspase 3 and chrome c caused by Ang II. According to these findings, simvastatin promoted mitophagy and inhibited Ang II-induced apoptosis, providing new therapeutic pathways for the treatment of HF.
According to a recent study, acute or chronic cardiac illness and pathological processes are mediated by lysosomal-regulated lipophagy, which is crucial for ATP generation and supply in cardiomyocytes [1]. The effects of simvastatin on lysosomal regulation and mitochondrial quality control It could be necessary to look into lipophagy more thoroughly, both in vitro and in vivo.
As summarized in Figure 7, our findings identified simvastatin as a potential mediator of mitochondrial protection against Ang II damage in HF and demonstrated its ability to lower ROS generation, control LDs and lysosome levels to supply energy for maintaining m, control mitochondrial quality control to promote mitophagy, and prevent activation of mitochondria-regulated apoptosis.
References
- Settembre C, Ballabio A. Lysosome: regulator of lipid degrada- tion pathways. Trends Cell Biol. 2014; 24: 743-50.
- Sleijfer S, van der Gaast A, Planting AS, Stoter G, Verweij J. The potential of statins as part of anti-cancer treatment. Eur J Cancer. 2005; 41: 516-22.
- Takayama N, Kai H, Kudo H, Yasuoka S, Mori T, Anegawa T, et al. Simvastatin prevents large blood pressure variabil- ity induced aggravation of cardiac hypertrophy in hypertensive rats by inhibiting RhoA/Ras–ERK pathways. Hypertens Res. 2011; 34: 341-7.
- Tsai JR, Chong IW, Chen YH, Hwang JJ, Yin WH, Chen HL, et al. Magnolol induces apoptosis via caspase- independent pathways in non-small cell lung cancer cells. Arch Pharm Res. 2014; 37: 548-57.
- Tsuruda T, Sekita-Hatakeyama Y, Hao Y, Sakamoto S, Kurogi S, Nakamura M, et al. Angiotensin II stimulation of car- diac hypertrophy and functional decompensation in osteoprotegerin- deficient mice. Hypertension. 2016; 67: 848-56.
- Ueta CB, Gomes KS, Ribeiro MA, Mochly-Rosen D, Ferreira JC. Disruption of mitochondrial quality control in peripheral artery disease: new therapeutic opportunities. Pharmacol Res. 2017; 115: 96-106.
- von Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol. 2017; 14: 323-41.
- Wang W, Fernandez-Sanz C, Sheu SS. Regulation of mitochondrial bioenergetics by the noncanonical roles of mitochondrial dynamics proteins in the heart. Biochimica et Biophysica Acta - Molecu- lar Basis of Disease. 2018; 1864: 1991-2001.
- Wang X, Yuan B, Dong W, Yang B, Yang Y, Lin X, et al. Humid heat exposure induced oxidative stress and apoptosis in cardiomyocytes through the Ang II signaling pathway. Heart Vessels. 2015; 30: 396-405.
- Wang Y, Chen B, Huang CK, Guo A, Wu J, et al. multiple murine models. JACC: Basic to Translational Science. 2018; 3: 503–517.
- Yan CH, Li Y, Tian XX, Zhu N, Song HX, Zhang J, et al. CREG1 ameliorates myocardial fibrosis associated with autoph- agy activation and Rab7 expression. Biochim Biophys Acta. 2015; 1852: 353-64.
- Yokoyama M, Seo T, Park T, Yagyu H, Hu Y, Son NH, et al. Effects of lipoprotein lipase and statins on cholesterol uptake into heart and skeletal muscle. J Lipid Res. 2007; 48: 646-55.
- Zablocki D, Sadoshima J. Ang II and oxidative stress in the fail- ing heart. Antioxid Redox Signal. 2013; 19: 1095-109.
- Zhang J, Yang Z, Xie L, Xu L, Xu D, Liu X. Statins, autophagy and cancer metastasis. Int J Biochem Cell Biol. 2013; 45: 745-52.
- Zhao W, Li Y, Jia L, Pan L, Li H, Du J. Atg5 deficiency mediated mitophagy aggravates cardiac inflammation and injury in response to Ang II. Free Radic Biol Med. 2014; 69: 108-15.
- Zhou L, Ma B, Han X. The role of autophagy in Ang IIinduced pathological cardiac hypertrophy. J Mol Endocrinol. 2016; 57: R143-52.
- Zou C, Qi H, Liu ZH, Han L, Zhao C, Yang X. Simvastatin activates the PPARγdependent pathway to prevent left ventricular hypertrophy associated with inhibition of RhoA signaling. Tex Heart Inst J. 2013; 40: 140-7.
- Settembre C, Ballabio A. Lysosome: regulator of lipid degrada- tion pathways. Trends Cell Biol. 2014; 24: 743-50.
- Sleijfer S, van der Gaast A, Planting AS, Stoter G, Verweij J. The potential of statins as part of anti-cancer treatment. Eur J Cancer. 2005; 41: 516-22.
- Takayama N, Kai H, Kudo H, Yasuoka S, Mori T, Anegawa T, et al. Simvastatin prevents large blood pressure variabil- ity induced aggravation of cardiac hypertrophy in hypertensive rats by inhibiting RhoA/Ras–ERK pathways. Hypertens Res. 2011; 34: 341-7.
- Tsai JR, Chong IW, Chen YH, Hwang JJ, Yin WH, Chen HL, et al. Magnolol induces apoptosis via caspase- independent pathways in non-small cell lung cancer cells. Arch Pharm Res. 2014; 37: 548-57.
- Tsuruda T, Sekita-Hatakeyama Y, Hao Y, Sakamoto S, Kurogi S, Nakamura M, et al. Angiotensin II stimulation of car- diac hypertrophy and functional decompensation in osteoprotegerin- deficient mice. Hypertension. 2016; 67: 848-56.
- Ueta CB, Gomes KS, Ribeiro MA, Mochly-Rosen D, Ferreira JC. Disruption of mitochondrial quality control in peripheral artery disease: new therapeutic opportunities. Pharmacol Res. 2017; 115: 96-106.
- von Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol. 2017; 14: 323-41.
- Wang W, Fernandez Sanz C, Sheu SS. Regulation of mito- chondrial bioenergetics by the noncanonical roles of mitochondrial dynamics proteins in the heart. Biochimica et Biophysica Acta Molecu- lar Basis of Disease. 2018; 1864: 1991-2001.
- Wang X, Yuan B, Dong W, Yang B, Yang Y, Lin X, et al. Humid heat exposure induced oxidative stress and apoptosis in cardiomyocytes through the Ang II signaling pathway. Heart Vessels. 2015; 30: 396-405.
- Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017; 120: 1812-24.
- Cao TT, Chen HH, Dong Z, Xu YW, Zhao P, Guo W, et al. Stachydrine protects against pressure overload-induced cardiac hypertrophy by suppressing autophagy. Cell Physiol Biochem. 2017; 42: 103-14.
- Choi SY, Park JS, Roh MS, Kim CR, Kim MH, Serebruany V. Inhibition of ang II-induced cardiac fibrosis by atorvastatin in adiponectin knockout mice. Lipids. 2017; 52: 415-22.
- Coelho-Filho OR, Shah RV, Mitchell R, Neilan TG, Moreno H, Simonson B, et al. Quantification of cardiomyocyte hypertrophy by cardiac magnetic resonance: implications for early cardiac remodeling. Circulation. 2013; 128: 1225-33.
- Coughlin L, Morrison RS, Horner PJ, Inman DM. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Invest Ophthalmol Vis Sci. 2015; 56: 1437-46.
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T et al. Mitochondrial oxidative stress mediates Ang II-induced cardiac hypertrophy and Gaq overexpression-induced heart failure. Circ Res. 2011; 108: 837-46.
- Delafontaine P, Akao M. Ang II as candidate of cardiac cachexia. Curr Opin Clin Nutr Metab Care. 2006; 9: 220-4.
- Delbosc S, Cristol JP, Descomps B, Mimran A, Jover B. Simvastatin prevents Ang II-induced cardiac alteration and oxidative stress. Hypertension. 2002; 40: 142-7.
- Domenighetti AA, Wang Q, Egger M, Richards SM, Pedrazzini T, Delbridge LM. Ang II-mediated phenotypic cardiomyocyte remodeling leads to age-dependent cardiac dysfunction and failure. Hypertension. 2005; 46: 426-32.
- Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, et al. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol. 2014; 24: 609-20.
- Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition. 2006; 22: 830-44.
- Gbelcová H, Svéda M, Laubertová L, Varga I, Vítek L, Kolár M, et al. The effect of simvastatin on lipid droplets accumula- tion in human embryonic kidney cells and pancreatic cancer cells. Lipids Health Dis. 2013; 12: 126.
- Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012; 15: 805-12.
- Goldenthal MJ. Mitochondrial involvement in myocyte death and heart failure. Heart Fail Rev. 2016; 21: 137-55.
- Griesser E, Vemula V, Raulien N, Wagner U, Reeg S, Grune T, et al. Cross-talk between lipid and protein carbonyla- tion in a dynamic cardiomyocyte model of mild nitroxidative stress. Redox Biol. 2017; 11: 438-55.
- Hwang KE, Kim YS, Jung JW, Kwon SJ, Park DS, Cha BK, et al. Inhibition of autophagy potentiates pemetrexed and simvastatin- induced apoptotic cell death in malignant mesothelioma and non-small cell lung cancer cells. Oncotarget. 2015; 6: 29482-96.
- Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000; 1486: 1-17.
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010; 160: 1577-9.
- Kuo HF, Liu PL, Chong IW, Liu YP, Chen YH, Ku PM, et al. Pigment epithelium-derived factor mediates autophagy and apoptosis in myocardial hypoxia/reoxygenation injury. PLOS ONE. 2016; 11: e0156059.
- Lee SJ, Zhang J, Choi AM, Kim HP. Mitochondrial dysfunc- tion induces formation of lipid droplets as a generalized response to stress. Oxid Med Cell Longev. 2013; 2013: 327167.
- Long Q, Yang K, Yang Q. Regulation of mitochondrial ATP syn- thase in cardiac pathophysiology. Am J Cardiovasc Dis. 2015; 5: 19-32.
- Losada MA, López A, Mateo J. Attenuation and diffusion pro- duced by small-radius curvatures in POFs. Opt Express. 2016; 24: 15710-20.
- Minami S, Yamamoto T, Takabatake Y, Takahashi A, Namba T, Matsuda J, et al. Lipophagy maintains energy homeosta- sis in the kidney proximal tubule during prolonged starvation. Autophagy. 2017; 13: 1629-47.
- Nishida K, Taneike M, Otsu K. The role of autophagic degrada- tion in the heart. J Mol Cell Cardiol. 2015; 78: 73-9.
- Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013; 62: 263-71.
- Pinchuk TV, Fedulaev YN, Khairetdinova GA, Denisova NN, Chura OV, Logunova IY. Anti-inflammatory effects of simva- statin in patients with chronic heart failure. Bull Exp Biol Med. 2014; 157: 552-4.
- Prathapan A, Vineetha VP, Raghu KG. Protective effect of Boerhaavia diffusa L. against mitochondrial dysfunction in Ang II induced hypertrophy in H9c2 cardiomyoblast cells. PLOS ONE. 2014; 9: e96220.
- Qin YW, Ye P, He JQ, Sheng L, Wang LY, Du J. Simvastatin inhibited cardiac hypertrophy and fibrosis in apolipoprotein E- deficient mice fed a ”Western-style diet” by increasing PPAR a and γ expression and reducing TC, MMP-9, and Cat S levels. Acta Pharmacol Sin. 2010; 31: 1350-8.
- Al-Rasheed NM, Al-Oteibi MM, Al-Manee RZ, Al-Shareef SA, Al-Rasheed NM, Hasan IH, et al. Simvastatin prevents isoproterenol-induced cardiac hypertrophy through modula- tion of the JAK/STAT pathway. Drug Des Dev Ther. 2015; 9: 3217-29.
- Aubdool AA, Thakore P, Argunhan F, Smillie SJ, Schnelle M, Srivastava S, et al. A novel acalcitonin gene-related peptide analogue protects against end-organ damage in experimental hypertension, cardiac hypertrophy, and heart failure. Circulation. 2017; 136: 367-83.
- Bhatt KN, Butler J. Myocardial energetics and heart failure: a review of recent therapeutic trials. Curr Heart Fail Rep. 2018; 15: 191-7.
- Björkhem-Bergman L, Lindh JD, Bergman P. What is a rele- vant statin concentration in cell experiments claiming pleiotropic effects? Br J Clin Pharmacol. 2011; 72: 164-5.
- Boovarahan SR, Kurian GA. Mitochondrial dysfunction: A key player in the pathogenesis of cardiovascular diseases linked to air pol- lution. Rev Environ Health. 2018; 33: 111-22.