
Review Article
Austin J Pharmacol Ther. 2023; 11(3):1178.
Can New Structure of RET Inhibitors Capable of Suppressing Resistant in Non-Small-Cell Lung Cancer?
Jiayi Shen¹; Liping Chen¹; Yulan Song¹; Sheng Chen²*; Wei Guo¹*; Yongdong Li¹*
¹Key Laboratory of Organo-Pharmaceutical Chemistry of Jiangxi Province, Gannan Normal University, Ganzhou, Jiangxi 341000, PR China
²Jiangxi Chiralsyn Biological Medicine Co., LTD, China
*Corresponding author: Wei Guo & Yongdong Li Key Laboratory of Organo-Pharmaceutical Chemistry of Jiangxi Province, Gannan Normal University, Ganzhou, Jiangxi 341000, PR China;
Sheng Chen, Jiangxi Chiralsyn Biological Medicine Co., LTD, China. Tel: +86 797 8393670; Fax: +86 797 8393670 Email: guoweigw@126.com; ydli2011@163.com; chensheng0813@163.com
Received: October 11, 2023 Accepted: November 18, 2023 Published: November 25, 2023
Abstract
In 2012, RET rearrangements are observed in 1-2% of Non-Small-Cell Lung Cancer (NSCLC) patients and result in the constitutive activation of downstream pathways normally implied in cell proliferation, growth, differentiation and survival. Several compounds have been reported, including some traditional kinases inhibitors and the discovery of some new structure of natural products. Cabozantinib and vandetanib are multikinase inhibitors have been explored in the clinic for NSCLC patients. As a result of the nonselective nature of these multikinase inhibitors, patients had off-target adverse effects. Then, the discovery and clinical validation of highly potent selective RET inhibitors such as pralsetinib and selpercatinib demonstrating improved effificacy and a more favorable toxicity profile. However, acquired resistance mediated by secondary mutations in the solvent-front region of the kinase (e.g. G810C/S/R) becomes a major challenge for selective RET inhibitor therapies. In this review, we will highlight typical RET inhibitors developed during these years and provide a reference for more potential RET inhibitors exploration in the future.
Keywords: REarranged during transfection (RET) kinase; Non-small cell lung cancer (NSCLC); Resistance; Inhibitors
Introduction
Lung cancer is the most common oncological disease, which is responsible for 11.6 % and 18.4 % of global cancer morbidity and mortality, respectively. It is classified for Small-Cell Lung Cancer (SCLC) and Non-Small Cell Lung Cancer (NSCLC). NSCLC is significantly more common than SCLC that accounts for about 85% and is further subdivided for squamous and non-squamous histological types [1]. Like other common NSCLC drivers, such as sensitizing Epidermal Growth Factor Receptor (EGFR) mutations and Anaplastic Lymphoma Kinase (ALK) or c-Rosproto-Oncogene 1 (ROS1) rearrangements, the oncogenic Rearranged during Transfection (RET) gene fusion was first identified in 2012 that was tend to occur in approximately 1-2 % of NSCLC and it was found to be more common in non-smoking or light smoking, young lung adenocarcinoma patients. RET gene was derived by DNA rearrangement during transfection of mouse NIH3T3 cells with human lymphoma DNA and located in the long arm of human chromosome 10 [2]. It encodes a receptor tyrosine kinase protein composed of 1143 transmembrane amino acid residues, and and consists of three regions. Up to now, 48 unique fusion partners in RET have been identified, such as KIF5B-RET, CCDC6-RET, and NCOA4-RET et al., [3]. These fusions lead to ligand-independent constitutive activation of the RET pathway and increased oncogenic signaling, resulting in RET gene overexpression. Interestingly, RET fusions were mutually exclusive with other oncogenic driver genes [4]. As patients harboring RET aberrations, selectively inhibiting the kinase is a promising therapeutic strategy [5].
For the treatment of NSCLC patients with RET alterations, several Multiple-targeted Kinase Inhibitors (MKIs) were approved [6,7]. Horeover, limited clinical benefits, relatively low tolerated doses, obvious adverse effects and mutations in the kinase prevent the broad application of these multiple-targeted drugs [8-14]. In 2020, two selective RET inhibitors, selpercatinib and pralsetinib were approved by US Food and Drug Administration (FDA). Several other highly promising selective RET inhibitors were also developed in different stages of clinical investigation [15-35]. However, acquired resistance conferred by secondary mutations were also identified. In this review, we focus on the present state of the RET inhibitors in the treatment of NSCLC, discuss the future perspectives for RET positive NSCLC patients and provide an updated panorama of this topic.
The Structure of RET
In 1985, Takahashi et al. [2] identified the protooncogene RET is a transforming gene located in the long arm of human chromosome 10 and was derived by DNA rearrangement during transfection of mouse NIH3T3 cells with human lymphoma DNA. The RET gene encodes a Receptor Tyrosine Kinase (RTK) protein composed of 1143 transmembrane amino acid residues and contains a large extracellular domain, a transmembrane domain and an intracellular tyrosine kinase domain [36]. The RET protein formed by in-frame fusion of the 5'-terminus of a chaperone gene with the 3'-terminus of RET containing its kinase structural domain [37]. The extracellular domain contains four Cadherin-Like Domains (CLD1-4), calcium binding site that between CLD2 and CLD3, a cysteine-richdomain and a conserved cysteinerich domain (Figure 1). Then, as the intracellular region contains a tyrosine kinase domain and tyrosine phosphorylation sites located next to the C terminal region. The C-terminal tail of RET has two major forms, which diverge after residue G1063 because of alternative splicing a short 9-amino acid one (RET9) and a long 51-amino acid one (RET51). Although the two isoforms share a largely common sequence and are coexpressed in many tissues, numerous studies have demonstrated differences in their temporal and spatial regulation of expression, cellular localization and trafficking and biologic functions. It has been suggested that RET51 is the more prominent isoform in tumors and it is more effective than RET9 at promoting cell proliferation, migration and anchorage-independent growth [38,39]. The combination of the intracellular kinase structural domain of RET and the coiled helix structural domain of the chaperone gene, leading to ligand-independent homodimerization and activation of RET by autophosphorylation tyrosine kinase, which in turn activates downstream pathways leading to tumorigenesis and development [36]. RET as the receptor is activated by the ligands and the function of the RET receptor which will be discussed as followed.
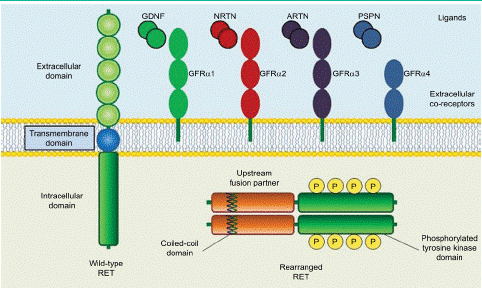
Figure 1: Structure of wild-type and rearranged RET proteins in a cancer cell.
The Functions of RET
In the decades that have passed since the discovery of RET, a lot of studies have clarified its function and biology and much has been uncovered related to its role in cancer. To date, three general mechanisms of aberrant RET activation have been reported in cancer. One of them is in-frame RET gene fusions [2,40], the other is targeted mutation of the RET gene itself [41-43] and the third one is aberrant overexpression of the RET gene [44,45]. These three mechanisms appear to share in common is the inappropriate activation of the tyrosine kinase, most commonly in the complete absence of ligand. In consideration of the RET ligands, including glial cell line derived neurotrophic factor (GDNF), neurturin, artemin and persephin, all belonging to the GDNF family (GFLs) [46]. These GFLs do not directly bind to RET and instead bind to GDNF family receptor-a (GFRa) coreceptors, which in turn recruit RET for dimerization [47,48]. Then, RET receptor is activated by a binary complex of glial cell GDNF family ligands with the coreceptor GDNF family receptor a(GFRa) [49,50]. GFL/GFRa/RET ternary complex triggers phosphorylation of the intracellular tyrosine residues and multiple downstream signaling activation including RAS/MAPK, PI3K/AKT and JAK/STAT pathways to regulate cell migration, proliferation and differentiation in physiological conditions [46,51] (Figure 2).
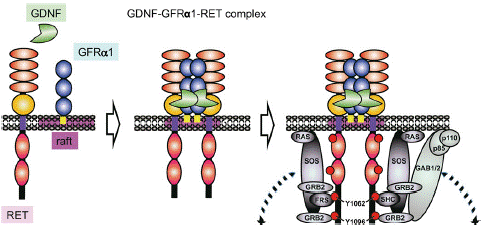
Figure 2: GDNF signaling via the GDNF-GFRa1-RET complex.
RET is activated in cancer mainly through chromosomal rearrangements that generate fusion genes containing the kinase domain of RET and gain-of-function missense mutations in both the extracellular and cytoplasmic regions of RET protein. Apart from these mechanisms, the increased expression level of wild-type RET has been linked to the pathogenesis of several cancer types.
RET plays important roles in the development of the kidney and nervous system. Studies in mouse models have shown that RET and the phosphorylation of its docking sites are critical for the growth and branching morphogenesis of ureteric bud cells from the metanephric mesenchyme [52,53]. RET is expressed in neural crest cells and required for the proliferation, differentiation, and survival of these cells [52,54]. RET is also involved in motoneuron survival and connectivity [55,56]. In addition, RET signaling contributes to the regulation and function of hematopoietic cells and spermatogenesis [57,58]. Loss-of-function RET mutations in humans have been linked to Hirschsprung disease, congenital anomalies of kidney or urinary tract, and congenital central hypoventilation syndrome [6]. RET gain-of-function alterations have been identified in multiple solid tumours. By sequencing more than 10,000 different metastatic tumours, RET alterations have been found in 2.4% of all cases, primarily in thyroid cancers and NSCLC. It is worth mentioning that, abnormal activation of RET mediated by mutation, overexpression, or rearrangement with other oncogenic partners are identified as driver forces in a variety of human malignancies [59-61]. The mutation and fusion of RET and it’s pathogenic factors will be discussed next.
RET Fusions
RET fusions are thought to be oncogenic for two reasons. First, fusion provides a mechanism to aberrantly express RET in a cell type where it is normally transcriptionally silent. Second, in all cases the extracellular domain is replaced with a protein dimerization domain. The outcome is the production of an intracellular RET tyrosine kinase domain capable of ligand-independent activation. RET fusions mainly identified as solid therapeutic targets, are found in 1-2% of NSCLC, implying that RET addicted malignancies are sensitive to targeted inhibition.
Testing for RET fusions is highly recommended in NSCLC, since it can predict benefits from targeted inhibition. Specifically, a variety of diagnostic tools are used to detect gene fusions at the DNA, RNA, and protein levels. The first RET fusion rearrangement and adenocarcinomas among NSCLC patients was identified by Kohno and Lipson and was reported in 2012 [3,62]. KIF5B-RET is the most frequent and the best characterized RET fusion, derived from a 10.6 Mb pericentric inversion on chromosome 10. CCDC6, NCOA4, and TRIM33 are also partner 5' genes for RET fusion in NSCLCs [63]. Until now, a total of 48 unique fusion partners in RET have been identified and at least 12 fusion RET partner genes have been identified in NSCLCs (Figure 3)
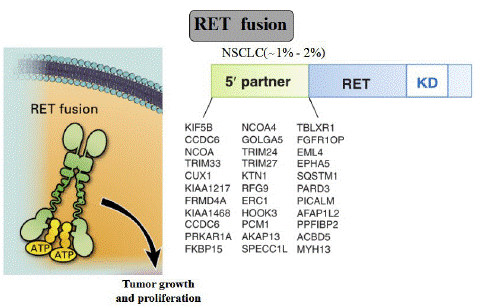
Figure 3: RET fusion.
The report revealed the prevalence of RET fusion genes was 1.8% in the overall population but among the 159 EGFR, ALK, ROS1, v-raf murine sarcoma viral oncogene homologue B1(BRAF), Kirsten Rat Sarcoma viral oncogene (KRAS), Human Epidermal growth factor Receptor 2 (HER2) WT patients, there was a 6.3% rate. Furthermore, Takeuchi et al. screened 1529 Japanese NSCLC patients, reporting RET chromosomal rearrangement in 0.9% of patients [64]. Thus, the prevalence of the RET fusion gene in lung adenocarcinoma is 0.9-1.8%. For all RET fusion variants, a chromosomal rearrangement leads to a blend between the coiled coil domain of the partner gene and RET intracellular kinase domain, whose function remains preserved despite the breakpoint. The coil coiled domain of the RET partner gene induces ligand independent homodimerization and activates the RET tyrosine kinase domain by auto-phosphorylation [65]. Among the downstream signals activated, preclinical models showed that RET fusion oncogenes (KIF5B-RET and CCDC6-RET) enhanced cell proliferation and survival via direct phosphorylation of STAT3 or activation of JAK/STAT3 and RAS/RAF/MEK/ERK signalling pathways [66]. It is also important to point out that key regulatory mechanisms of RET inactivation, such as endocytosis and recruitment of membrane associated ubiquitin ligases, do not appear to impact the fusion proteins, which additionally may enhance their oncogenicity [67,68]. Need to add that, the mechanism of activation of RET fusion proteins is analogous to the oncogenic activation of rearranged ALK in NSCLC, but clearly differs from ROS1. In the EML4-ALK fusion gene, a coiled-coil domain in EML4 is fused to the ALK kinase domain, conferring oligomerization and constitutive kinase activation [69], while coil-coiled domains are not consistently present in ROS1 fusion genes in NSCLC and they are not necessary to drive oncogenesis [64]. The tumorigenic potential of RET fusion proteins has been demonstrated in vitro in Ba/F3 (pro-B lymphocyte) [3] or NIH3T3 (fibroblast) cell lines [62,64], and in CCDC6-RET-positive LC-2 lung adenocarcinoma cells [70,71]. Furthermore, the transforming potential of RET fusion gene was also evaluated in vivo, in athymic mice through subcutaneous injection of KIF5B-RET transfected NIH3T3 cells [64], and in transgenic immunocompetent KIF5B-RET-rearranged mice [18,58]. Of note, in the latter in vivo models, after tumour development, continuous KIF5B–RET fusion gene expression was required for lung tumour survival. Furthermore, RET-rearranged lung adenocarcinoma in transgenic mice presented a strong desmoplastic reaction and aggressive features [72].
In addition, RET fusions are often present in NSCLC patients without other oncogenic drivers. Thus, the clinical and pathological characteristics of RET+ patients may differ from what has been observed for those with other oncogenic drivers. Moreover, some initial reports showed that lymphangitic spread and psammoma bodies were frequently reported in a small series of RET-rearranged NSCLC, suggesting that RET assessment should be encouraged in those cases as mentioned [73].
As RET was first identified more than 10 years ago, which is activated in cancer mainly through chromosomal rearrangements that generate fusion genes containing the kinase domain of RET, implying that RET addicted malignancies are sensitive to targeted inhibition. With the study of RET in NSCLC, clinical treatments and inhibitors of it were gradually from bench to bedside, especially opening the door for small molecule inhibitors. We will discuss the multikinase inhibitors (MKIs), the selective inhibitors and some other reported inhibitors as follows.
RET Inhibitors
Multiple-Targeted Kinase Inhibitors
Cabozantinib: The first glimmer of hope for the patients with RET-rearranged NSCLC came with the discovery of multikinase inhibitors (MKIs) (Figure 4) The first multitarget inhibitors of RET we discuss is cabozantinib (1), which was approved by the US FDA in 2016. Cabozantinib (XL-184), with the structure of N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, was developed by Exelixis Inc. The drug has low nanomolar activity against RET (the IC50 for RET is 5.2 nM), and it also has activity against ROS1, MET, VEGFR2, AXL, TIE2, and vkit Hardy-Zuckerman 4 feline sarcoma viral oncogene homologue (KIT) [74].
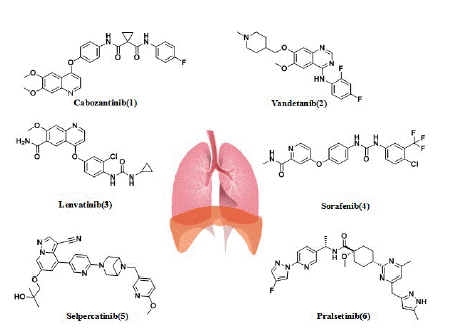
Figure 4: Multikinase inhibitors and RET-selective inhibitors in patients with RET-positive lung cancer.
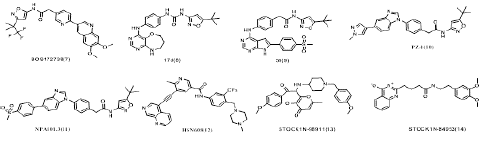
Figure 5: Chemical structures of several other selective RET inhibitors.
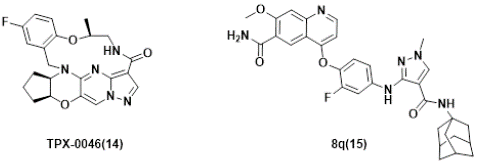
Figure 6: Several RET inhibitors suppressing resistant mutants in solvent-front regions.
Drilon et al. evaluated the safety and activity of cabozantinib in 26 patients with advanced RET-rearranged NSCLC in an open-label, single-arm, phase II trial [8]. It was the first study of cabozantinib to demonstrate the activity of a RET inhibitor in a molecularly enriched cohort of patients with advanced-stage, RET-rearranged NSCLC [8,75]. In this trial, the primary objective was to determine the overall response, and secondary outcomes included Progression-Free Survival (PFS), Overall Survival (OS), and safety. The Overall Response Rate (ORR) was 28%, with 7 of 25 evaluable patients achieving a partial response, including patients with KIF5B–RET, TRIM33–RET, and CLIP1–RET fusions. The median progression-free (mPFS) survival was 5.5 months, and median overall survival was 9.9 months.
In this disease context, this trial was followed by two phase II trials of vandetanib, one conducted in Japan (LURET) [76] and the other one in South Korea [9] ,which will be discussed next. The common adverse reactions caused by cabozantinib include abdominal complex signs of diarrhea, cavity inflammation, Palmar-Plantar Erythrodysesthesia Syndrome (PPES), body mass reduction, appetite lethargy, nausea, fatigue, oral pain, hair color change, taste disturbance, high blood pressure, abdominal pain and constipation [77,78].
Vandetanib: Vandetanib (ZD6474) is a 4-anilinoquinazoline-like drug molecule, with the structure of N-(2,4-difluorophenyl)-6-methoxy-7-((1-methylpiperidin-4-yl)methoxy) quinazoline-4 amine(2), which was developed by AstraZeneca (UK) and approved by the US FDA in 2011. Vandetanib is an orally active low-molecular multitargeted tyrosine kinase inhibitor with activity against EGFR, VEGFR-2 and RET(the IC50 for RET is 100 nM) [79]. Preclinical studies demonstrated the antitumor activity of vandetanib both in vitro and in vivo against LC-2/ad cells carrying the CCDC6-RET fusion [70]. As mentioned above, in Japan (LURET) phase II study, the enrolled 19 Japanese RET fusion patients received the vandetanib treatment, the mPFS was 4.7 months, the median OS was 11.1 months, and the OS at 12 months was 52.6%. Eleven patients (57.9%) had adverse events leading to a dose reduction [38]. Another phase II study explored the efficacy of vandetanib in Korean patients with metastatic or recurrent RET fusion NSCLC. This study showed the mPFS was 4.5 months, the median OS was 11.6 months. The most common grade 3 adverseevents (AEs) were hypertension (17%), a prolonged QTc interval (11%), and transaminitis (6%) [80].
In the clinical trial of vandertanil for NSCLC described above, patients died most are caused by disease progression, but there are also side effects that cannot be tolerated discontinue the medication. Common side effects were diarrhea, rash, hypertension, and asymptomatic prolonged QT interval, nausea, vomiting, neutropenia, anemia, fatigue, etc. But most can be tolerated or can be relieved after symptomatic treatment. Also, the adverse reaction was higher of Vandertanil combined application [81-84].
Lenvatinib: Lenvatinib (E7080) with the structure of 4-(3-chloro-4-(3-cyclopropylureido)phenoxy)-7-methoxyquinoline-6-carboxamide(3) is a multitargeted tyrosine kinase inhibitor of VEGFR1-3, fibroblast growth factor receptors (FGFR)1-4, platelet-derived growth factor receptor alpha (PDGFRa), KIT, and RET(the IC50 for RET is 1.5 nM) [87-90]. It was developed by Eisai Inc. and was approved by the US FDA in 2015 [87]. In a phase II trial, lenvatinib (24 mg/d) was tested in 25 patients with RET-rearranged NSCLC. Of them, 52% had a KIF5B-RET rearrangement and 48% had different known RET fusion genes. Interestingly, 28% of patients received lenvatinib after a previous line of anti-RET therapy. The ORR, Disease control rates (DCR), and mPFS times were 16%, 76%, and 7.3 months, respectively. In seven patients who had received previous RET therapy, ORR with lenvatinib was superimposable (14%) on the response seen in RET TKI-naive patients. Although the ORR was equivalent (15%-17%) in patients with the KIF5B-RET rearrangement and in patients with different known RETS fusion genes, the mPFS was lower in patients with the KIF5B-RET rearrangement than in patients with other known fusion variants (3.6 versus 9.1 months). Lenvatinib induced grade 3 to 4 AEs in 92% of the patients (hypertension in 58% and proteinuria in 16%); dose reduction and drug discontinuation occurred in 64% and 76% of patients, respectively. Lenvatinib also has side effects like high blood pressure, diarrhea, and thrombocytopenia [89].
Sorafenib: Sorafenib (BAY 43-9006) with the structure of 4-(4-(3-(4-chloro-3-(trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide(4), targets VEGFR1, VEGFR2, VEGFR3, platelet derived growth factor receptor beta (PDGFRB), c-KIT, Fms-Like Tyrosine kinase 3 (FLT3), and also RET (the IC50 for RET inhibition was 15-150 nM) [81]. It was developed by Bayer and was approved by the US FDA in 2005. In vitro, sorafenib suppressed the growth of KIF5B-RET-transfected Ba/F3 pro-B lymphocytes [3]. In vivo, the efficacy of sorafenib has been tested in a limited number of patients (n=3) in a study by Horiike et al. [89] One patient experienced Stable Disease (SD) while two showed Progressive Disease (PD) as best responses to treatment. However, the antitumor activity of sorafenib does not appear to be significant. The most common side effects were palmar metatarsal syndrome, hypertension, and diarrhea [90].
Whereas multikinase inhibitors are active in patients with RET-driven NSCLCs, response rates achieved in prospective series are lower than those observed in other driver-positive, advanced-stage tumours with matched targeted therapies. One possible explanation for the limited efficacy of RET-directed therapy with multikinase inhibitors relates to the inhibition of non-RET kinases, as well as non-kinase targets. It has been validated that selectively inhibiting the kinase is a promising therapeutic strategy for patients harboring RET aberrations. Two highly potent and selective RET TKIs, selpercatinib and pralsetinib have been developed and their activity has been investigated, which will be discussed below.
Selective RET Inhibitors
Small, highly selective RET inhibitors have been developed with the aim of overcoming treatment-related toxicities commonly seen with non-selective RET inhibitors. Among these, selpercatinib and pralsetinib received FDA approval for the treatment of NSCLC harbouring RET alterations.
Selpercatinib
Selpercatinib (LOXO-292) with the structure of 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-((6-methoxypyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile(5) has been developed as a selective, ATP-competitive RET-inhibitor (the IC50 for RET is 2.0 nM), which was developed by Lilly. Selpercatinib is a highly selective RET inhibitor because it could block the adenosine triphosphate binding site of RET receptor tyrosine kinase [91]. Data on its preclinical characterization and activity was published in 2018 [92]. On May 8th of 2020, the US FDA approved selpercatinib as the first targeted therapy for RET-rearranged NSCLC [93].
The approval for selpercatinib was based on the ORR with prolonged duration of responses seen in a multicenter, open-label, multicohort clinical trial (LIBRETTO-001, NCT03157128)[94]. The accelerated approval of selpercatinib was based on the benefit-risk evaluation of the results of LIBRETTO-001. After that, the efficacy of selpercatinib in RET fusion-positive NSCLC was evaluated in 105 patients previously treated with platinum chemotherapy and 39 treatment-naive patients. ORRs for previously treated and treatment-naive patients were 64% and 85%, respectively. Patients with RET-mutant MTC were also divided into two cohorts: a) previously treated with cabozantinib or vandetanib (N=55) and b) cabozantinib and vandetanib naive (N=88). ORRs for the cohorts were 69% and 73%, respectively. The trial is still active and intends to further study the clinical benefits of selpercatinib [95].
In 2021, selpercatinib was approved by the European Medicines Agency (EMA) and Swiss-Medic for second line or posterior line therapy. The clinical trial LIBRETTO-321 was conducted to evaluate the efficacy of selpercatinib for Chinese RET fusion NSCLC patients. The ORR was 61.1% in the selpercatinib treated population. About 90% of the patients remained in continuous remission after 6 months [96]. This study indicated that selpercatinib was also a promising therapeutic option for Chinese RET fusion NSCLC patients. Common AEs with selpercatinib included increased glutamic transaminase (AST) levels (51%), increased Alanine aminotransferase (ALT) levels (45%), dry mouth (39%), diarrhea (37%), hypertension (35%) and rash (27%), and most of the AEs were grade 1 or 2 [96].
Pralsetinib: Pralsetinib (BLU-667) with the structure of (1s,4R)-N-((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)methyl)pyrimidin-2-yl)cyclohexane-1-carboxamide(6), is an oral Tyrosine Kinase Inhibitors (TKI) with potent and specifific activity against the RET kinase domain(the IC50 for RET inhibition was 0.4 nM), including multiple RET alterations such as fusions, activating point mutations and predicted acquired resistance mutations, which was developed by Blueprint Medicines Corporation.
In vitro studies demonstrated that, compared with cabozantinib and vandetanib, pralsetinib is 8 to 28-fold more potent against the wild-type RET kinase domain. Moreover, pralsetinib also displays a strong activity against common oncogenic RET alterations, such as RET M918T, KIF5B–RET and CCDC6–RET fusions [97].
The clinical activity and safety of pralsetinib was investigated by the ARROW study (Global multicentric single-arm phase I/II trial) [98]. Based on the result of this study, pralsetinib was approved as first-line or post-line treatment for RET fusion NSCLC by FDA in September 2020 [99,100]. Updated data reported by the American Society of Clinical Oncology(ASCO) in 2021 showed that the ORR was 17.1 months, the CR was 6%, and the mPFS was 16.5 months (n=136). Nine patients with measurable brain metastases all showed an intracranial reduction to a certain extent (intracranial Response Rate (RR) 56%, intracranial Complete Remission (CR) 33%). It was also demonstrated that pralsetinib had signifificant intracranial activity. As the excellent effificacy and low off-target toxicity in RET cancer patients, pralsetinib was also approved by China’s State Food and Drug Administration (NMPA) in March, 2021 [101]. This is the first RET inhibitor approved in China and is of great signifificance [94,102,103].
In terms of side effects, pralsetinib has been well tolerated with mainly low grade toxicities (28% had = grade 3 events). The most commonly observed adverse events were AST and ALT increase (22% and 17%, respectively), hypertension (18%), constipation (17%), neutropenia (15%) and fatigue (15%) [104].
Other RET inhibitors in development: Except for selpercatinib and pralsetinib, BOS172738/Zeteletinib (Phase I, NCT03780517)[105,106], GSK3179106 (phase I, NCT02727283) [107], SY-5007 (phase I, NCT05278364), KL590586 (phase I/II, NCT05265091) and HS-10365 (phase I, NCT05207787), were also developed in different stages of clinical investigation. For example, BOS172738(7) is a targeted inhibitor of aberrant mutations in RET. A phase I clinical trial of BOS172738 reported that BOS172738 showed good safety for long-term administration. The overall efficacy ORR assessed by the investigator was 33% (n=18/54), and the NSCLC cohort ORR was 33% (n=10/30) [108,109]. Currently, multiple clinical trials are being conducted, including LIBRETTO-431, LIBRETTO-531, NCT04211337, and NCT03780517 [110].
In addition, N-phenyl-7,8-dihydro-6H-pyrimido[5,4-b][1,4]oxazin-4-amine derivatives have been reported as a new class of RET inhibitors and one of the representative compounds 17d(8) [35], 1-(5-(tert-butyl)isoxazol-3-yl)-3-(4-((6,7,8,9-tetrahydropyrimido[5,4-b][1,4]oxazepin-4-yl)amino)phenyl)urea, potently inhibits RET(the IC50 for RET is 10 nM) and its drug resistance mutants RET-V804M and RET-V804L. Lakkaniga et al. [24] investigated a series of pyrrolo[2,3-d]pyrimidine-based derivatives and identified a lead compound, named 59(9), is a type II inhibitor of RET, which shows low nanomolar potency against RET(the IC50 for RET is 6.8 nM) and RETV804M (the IC50 for RET is 13.5 nM) and additionally proposed a binding pose of this compound in RET pocket. The group of Moccia et al. [20] identified the clinical drug candidates Pz-1(10) and NPA101.3(11) with the IC50 for RET is less than 1.0 nM and 1.0 nM respectively. Interestingly, Pz-1(10) and NPA101.3(11) lacking the structural liability for demethylation showed a selective inhibitory profile for both VEGFR2 and RET (WT and V804M).
What is more, during the past 10 years, Wang et al.[80] synthesized various nicotinamide analogs based on the scaffold of benzamide aminonaphthyridine HSN356, which was reported to inhibit RET kinase [111]. HSN608(12), the nicotinamide analog of HSN356 exerts strong RET inhibition and also inhibit RET(V804M/L) and RET(S905F) mutants better than alectinib, sorafenib, vandetanib and apatinib, and comparable to pralsetinib.
RET inhibitors was also discovered by virtual screening of Natural Product (NP) libraries, four natural product (NP) libraries encompassing Otava NP , NPASS (Natural Product Activity & Species Source) NP, IBS (InterBioScreen) NP , and LC (Life Chemicals) NP were screened using the produced model. The model as a 3D query was employed to screen four natural product (NP) libraries, including a total of 102,829 NPs. Subsequent virtual screening procured 198 compounds, which were subjected to computer-aided drug designing (CADD). Among the investigated candidates, STOCK1N-98911 (13) and STOCK1N-84953 (14) exhibited favorable interaction energies towards RET and another kinases [112]. Even the compounds did not have the activity as much as other inhibitors, but it show us another way to discovery new structure RET inhibitors.
Despite the significant progress and promising clinical outcomes of the selective RET kinases inhibitors, acquired resistance conferred by secondary mutations, e.g. G810C/S/R in Solvent-Front region (SF), Y806 C/N (in hinge residue) or V738A (in β2 strand) [96,113], was identified. Consequently, substantial efforts have been devoted to discover new generation selective RET inhibitors for combating unsolved clinical needs [114,115], and at least 4 candidates have been advanced into clinical trials. Examples include TPX-0046 (Phase I/II, NCT04161391) [116,117], LOXO-260 (Phase I, NCT05241834) [118] and TAS0953/HM06 (phase I/II, NCT04683250) [119], Investigation New Drug (IND) was also filed for APS03118 (undisclosed structure) based on the company’s announcement [120], which will be discussed next.
TPX-0046 with a small and rigid macrocyclic structure of (13E,14E,15aR,18aS,5S)-35-fluoro-5-methyl-15,15a,16,17,18,18a-hexahydro-4-oxa-7-aza-1(5,3)-cyclopenta[b]pyrazolo[1',5':1,2]pyrimido[4,5-e][1,4]oxazina-3(1,2)-benzenacyclooctaphan-8-one(14), is developed by Turning Point Therapeutics. It is a potent and selective next-generation orally bioavailable RET/SRC kinase inhibitor with a small and rigid macrocyclic structure that is structurally differentiated from current RET inhibitors. In enzymatic assays, TPX-0046 demonstrated low nanomolar potency against WT and many mutated RETs, as well as SRC. which is VEGFR2-sparing. TPX-0046 potently inhibited RET phosphorylation and cell proliferation in in-house engineered Ba/F3 KIF5B-RET, TT, and LC2/ad cells with IC50 of approximately 1nM. TPX-0046 is also potent against the Solvent Front Mutations (SFM) G810R in Ba/F3 cell proliferation assay with a mean IC50 of 17 nM, whereas comparable proxy molecules for pralsetinib and selpercatinib have IC50 >500 nM. TPX-0046 demonstrated marked anti-tumor efficacy in vivo in multiple RET-driven cancer cell-derived and patient-derived xenograft tumor models [116]. The clinical trial (ClinicalTrials.gov Identifier: NCT04161391) employing TPX-0046 is underway [121,122].
However, other compounds like LOXO-260 (Phase I, NCT05241834) [120], TAS0953/HM06 (phase I/II, NCT04683250) [121] and APS03118 which is filed by Investigation New Drug (IND) haven’t shown any clinical data until now. The only thing about APS03118 is the IC50 for mutant RET inhibition was less than 0.4 nM) which is based on the company’s announcement, but the structure of it was undisclosed [122].
Lately, Ding’s group [123], reported a structure-based design of 1-methyl-3-((4-(quinolin-4-yloxy)phenyl)amino)-1H-pyrazole-4-carboxamide derivatives recently. One of the representative compounds, named 8q(15), potently suppressed wild-type RET kinase with an IC50 value of 13.7 nM. It also strongly inhibited the proliferation of BaF3 cells stably expressing various oncogenic fusions of RET kinase with solvent-front mutations , e.g. CCDC6-RETG810C, CCDC6-RETG810R, KIF5BRETG810C and KIF5B-RETG810R, with IC50 values of 15.4, 53.2, 54.2 and 120.0 nM, respectively. Furthermore, it also dose-dependently inhibited the activation of RET and downstream signals and obviously triggered apoptosis in Ba/F3-CCDC6-RETG810C/R cells. The compound also exhibited significant anti-tumor efficacy with a Tumor Growth Inhibition (TGI) value of 66.9% at 30 mg/kg/day via i. p. in a Ba/F3-CCDC6-RET G810C xenograft mouse model.
Nevertheless, no drug is approved for overcoming acquired resistance against the 2nd generation selective RET inhibitor therapies to date. As metioned, some mutation occured in RET-positivite NSCLCs, we will discuss next.
RET Mutations
It was an intraget kinase-acquired resistance that dynamically evolves under kinase inhibitor selection pressure, making the kinase continuously activated under medication conditions. Gatekeeper mutations and solvent-front mutations were included. It has been reported that resistance mechanisms in MKIs include RET V804M gatekeeper mutations and RET S904F [113]. In RET positive NSCLS patients, the primary V804 M/L/E and S904F mutations in the kinase gatekeeper and activation loop, respectively, formed steric clashes with the drugs [124-126].
The selective RET inhibitor selpercatinib and pralsetinib induced a pre-lytic mutation (G810A/S) [127]. It also demonstrated that it increased kinase activity and conferred resistance through allosteric effects. Also, selective RET inhibitors have been designed to overcome gatekeeper mutations. The concurrent RET V804M gatekeeper mutation was associated with a G810 resolute mutation in an NSCLC patient.
The selective RET inhibitor selpercatinib and pralsetinib induced a pre-lytic mutation (G810A/S) [126]. It also demonstrated that it increased kinase activity and conferred resistance through allosteric effects. Also, selective RET inhibitors have been designed to overcome gatekeeper mutations. The concurrent RET V804M gatekeeper mutation was associated with a G810 resolute mutation in an NSCLC patient.
For example, RET mutations located at the floor of the solvent-front (G810C/S/R), the hinge (Y806C/N), and the β2 strand (V738A) of the RET ATP-binding site [95,108,113] in addition to target-by pass mechanisms [108,128,129]. Among these mutations, the G810C/S/R mutations displayed the strongest resistance[95] and were observed more often in patients whose tumors developed resistance to selpercatinib. Selpercatinib-resistant RET mutations identified so far were cross-resistant to pralsetinib [95]. Interestingly, Wu’s group reported that the L730V/I mutations at the roof of the solvent-front site of the RET kinase were strongly resistant to pralsetinib but not to selpercatinib [130].
Conclusions and Perspectives
NSCLC takes a leading position with regard to recent improvements in life expectancy as compared to other common tumor types, the use of appropriate targeted drugs results in manifold increase of their overall survival. The development of mutation-tailored drugs is reaching some plateau, particularly, some kinase inhibitors that are focus on suppressing resistant mutants in solvent-front regions. As NSCLC exome sequencing studies did not reveal significant number of novel potentially druggable targets. With the increasing demand for combining the expertize in molecular biology, pathology and clinical as pects of cancer management, the front-line of integration of clinical research, NSCLC may serve as an example of precision medicine, which will allow development of ideal treatment strategies for RET positive NSCLC patients.
In the past decades RET oncogene has emerged as a critical tumorigenesis driver. RET mutations and rearrangements now represent a well-established mechanism that drives tumor growth across several types of neoplasms, including thyroid and lung cancer. Treatment with non-specific MKIs in RET fusion-positive NSCLC achieved modest clinical outcomes and limited response durability, especially when compared with those achieved by targeting oncogenic drivers other than RET. Therefore, the two highly selective RET inhibitors, pralsetinib and selpercatinib, were specifically developed to target RET kinases selectivity and to overcome resistances to MKIs. These compounds have received FDA breakthrough designation and have been approved for clinic use based on the results of the LIBRETTO-001 and ARROW trials. Although these agents have been developed to overcome MKIs limits and have demonstrated remarkable clinical activity, new mechanisms of acquired resistance have already been reported. The emergence of off-target RET-independent mechanisms of resistance to pralsetinb and selpercatinib has highlighted the necessity to test further next-generation agents and to explore new therapeutic strategies, including concurrent inhibition of RET and parallel signaling pathways of resistance. What’s more, the treatment of two selective RET inhibitors costs are high. For example, selpercatinib is priced at $20,600 a month. It is difficult for the average patients to benefit.
Within the next decade, the field of RET inhibition in NSCLC is on the verge of a breakthrough that will give physicians and patients promising new therapeutic options. Identifying potent, selective, and less toxic RET target agents, looking for compounds with RET activity from natural products, exploring the potential impact of different fusion variants, characterizing concomitant molecular alterations and mechanisms of resistance to RET inhibition to identify optimal therapeutic combinations represent the challenges for future research in this field of NSCLC treatment.
Author Statements
Author Contributions
S.J. and C.L. wrote the paper; C.S. and G.W. checked the paper; S.Y. and L.Y. gave advice. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (No. 82360675), Education Department of Jiangxi Province (GJJ211439), Ganzhou Administration of Science & Technology (No. 202101124696) and the Doctor Foundation of Gannan Normal University (No. BSJJ202106).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68: 394-424.
- Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985; 42: 581-8.
- Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012; 18: 382-4.
- Wang R, Hu HC, Pan YJ, Li Y, Ye T, Li CG, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012; 30: 4352-4358.
- Lu C, Zhou Q. Diagnostics, therapeutics and RET inhibitor resistance for RET fusion-positive non-small cell lung cancers and future perspectives. Cancer Treat Rev. 2021; 96: 102153.
- Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. 2018; 15: 151-67.
- Subbiah V, Yang D, Velcheti V, Drilon A, Meric-Bernstam F. State-of-the-art strategies for targeting RET-dependent cancers. J Clin Oncol. 2020; 38: 1209-21.
- Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016; 17: 1653-60.
- Lee SH, Lee JK, Ahn MJ, Kim DW, Sun JM, Keam B, et al. Vandetanib in pretreated patients with advanced non-small cell lung cancer harboring RET rearrangement: a phase II clinical trial. Ann Oncol. 2017; 28: 292-7.
- Hida T, Velcheti V, Reckamp KL, Nokihara H, Sachdev P, Kubota T, et al. A phase 2 study of lenvatinib in patients with RET fusion-positive lung adenocarcinoma. Lung Cancer. 2019; 138: 124-30.
- Gupta-Abramson V, Troxel AB, Nellore A, Puttaswamy K, Redlinger M, Ransone K, et al. Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol. 2008; 26: 4714-9.
- Wells Jr SA, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012; 30: 134-41.
- Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013; 31: 3639-46.
- Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014; 384: 319-28.
- Cincinelli R, Cassinelli G, Dallavalle S, Lanzi C, Merlini L, Botta M, et al. Synthesis, modeling, and RET protein kinase inhibitory activity of 3- and 4-substituted β-carbolin-1-ones. J Med Chem. 2008; 51: 7777-87.
- Mathison CJN, Yang Y, Nelson J, Huang Z, Jiang J, Chianelli D, et al. Antitarget selectivity and tolerability of novel pyrrolo[2,3-d]pyrimidine RET inhibitors. ACS Med Chem Lett. 2021; 12: 1912-9.
- Newton R, Waszkowycz B, Seewooruthun C, Burschowsky D, Richards M, Hitchin S, et al. Discovery and optimization of wt-RET/KDR-selective inhibitors of RET kinase. ACS Med Chem Lett. 2020; 11: 497-505.
- Drilon A, Fu S, Patel MR, Fakih M, Wang D, Olszanski AJ, et al. A phase I/Ib trial of the VEGFR-sparing multikinase RET inhibitor RXDX-105. Cancer Discov. 2019; 9: 384-95.
- Frett B, Carlomagno F, Moccia ML, Brescia A, Federico G, De Falco V, et al. Fragment-based discovery of a dual pan-RET/VEGFR2 kinase inhibitor optimized for single-agent polypharmacology. Angew Chem Int Ed Engl. 2015; 54: 8717-21.
- Moccia M, Frett B, Zhang L, Lakkaniga NR, Briggs DC, Chauhan R, et al. Bioisosteric discovery of NPA101.3, a second generation RET/VEGFR2 inhibitor optimized for single-agent polypharmacology. J Med Chem. 2020; 63: 4506-16.
- Mathison CJN, Chianelli D, Rucker PV, Nelson J, Roland J, Huang Z, et al. Efficacy and tolerability of pyrazolo[1,5-a]pyrimidine RET kinase inhibitors for the treatment of lung adenocarcinoma. ACS Med Chem Lett. 2020; 11: 558-65.
- Yoon H, Kwak Y, Choi S, Cho H, Kim ND, Sim T. A pyrazolo[3,4-d]pyrimidin-4-amine derivative containing an isox-azole moiety is a selective and potent inhibitor of RET gatekeeper mutants. J Med Chem. 2016; 59: 358-73.
- Luo Z, Wang L, Fu Z, Shuai B, Luo M, Hu G, et al. Discovery and optimization of selective RET inhibitors via scaffold hopping. Bioorg Med Chem Lett. 2021; 47: 128149.
- Lakkaniga NR, Gunaganti N, Zhang L, Belachew B, Frett B, Leung YK, et al. Pyrrolo[2,3-d]pyrimidine derivatives as inhibitors of RET: design, synthesis and biological evaluation. Eur J Med Chem. 2020; 206: 112691.
- Toenjes ST, Garcia V, Maddox SM, Dawson GA, Ortiz MA, Piedrafita FJ, et al. Leveraging atropisomerism to obtain a selective inhibitor of RET kinase with secondary activities toward EGFR mutants. ACS Chem Biol. 2019; 14: 1930-9.
- Dinér P, Alao JP, Söderlund J, Sunnerhagen P, Grøtli M. Preparation of 3-substituted-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amines as RET kinase inhibitors. J Med Chem. 2012; 55: 4872-6.
- Moccia M, Liu Q, Guida T, Federico G, Brescia A, Zhao Z, et al. Identification of novel small molecule inhibitors of oncogenic RET kinase. PLOS ONE. 2015; 10: e0128364-e0128376.
- Abdel-Magid AF. RET kinase inhibitors may treat cancer and gastrointestinal disorders. ACS Med Chem Lett. 2015; 6: 13-4.
- Xu Y, Gao C, Andreasson M, Håversen L, Carrasco MP, Fleming C, et al. Design and development of photoswitchable DFG-Out RET kinase inhibitors. Eur J Med Chem. 2022; 234: 114226.
- Li X, Su J, Yang Y, Lian W, Deng Z, Yang Z, et al. Discovery of 4-methyl-N-(4-((4-methylpiperazin-1-yl)methyl-3-(trifluoromethyl)phenyl)-3-((6-(pyridin-3-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)-oxy)benzamide as a potent inhibitor of RET and its gatekeeper mutant. Eur J Med Chem. 2020; 207: 112755-112776.
- La Pietra V, Sartini S, Botta L, Antonelli A, Ferrari SM, Fallahi P, et al. Challenging clinically unresponsive medullary thyroid cancer: discovery and pharmacological activity of novel RET inhibitors. Eur J Med Chem. 2018; 150: 491-505.
- Newton R, Bowler KA, Burns EM, Chapman PJ, Fairweather EE, Fritzl SJR, et al. The discovery of 2-substituted phenol quinazolines as potent RET kinase inhibitors with improved KDR selectivity. Eur J Med Chem. 2016; 112: 20-32.
- Frett B, Moccia M, Carlomagno F, Santoro M, Li HY. Identification of two novel RET kinase inhibitors through MCR-based drug discovery: design, synthesis and evaluation. Eur J Med Chem. 2014; 86: 714-23.
- Yoon H, Shin I, Nam Y, Kim ND, Lee KB, Sim T. Identification of a novel 5-amino-3-(5-cyclopropylisoxazol-3-yl)-1-isopropyl-1H-pyrazole-4-carbox-amie as a specific RET kinase inhibitor. Eur J Med Chem. 2017; 125: 1145-55.
- Yang J, Chen K, Zhang G, Yang Q-Y, Li Y-S, Huang S-Z, et al. Structural optimization and structure-activity relationship stu-dies of N-phenyl-7,8-dihydro-6H-pyrimido oxazin-4-amine derivatives as a new class of inhibitors of RET and its drug resistance mutants. Eur J Med Chem. 2018, 143: 1148-64.
- Ferrara R, Auger N, Auclin E, Besse B. Clinical and translational implications of RET rearrangements in non-small cell lung cancer. J Thorac Oncol. 2018; 13: 27-45.
- Takahashi M, Buma Y, Iwamoto T, Inaguma Y, Ikeda H, Hiai H. Cloning and expression of the RET proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene. 1988; 3: 571-8.
- Rossel M, Pasini A, Chappuis S, Geneste O, Fournier L, Schuffenecker I, et al. Distinct biological properties of two RET isoforms activated by MEN 2A and MEN 2B mutations. Oncogene. 1997; 14: 265-75.
- Lian EY, Maritan SM, Cockburn JG, Kasaian K, Crupi MJ, Hurlbut D, et al. Differential roles of RET isoforms in medullary and papillary thyroid carcinomas. Endocr Relat Cancer. 2017; 24: 53-69.
- Takahashi M, Cooper GM. Ret transforming gene encodes a fusion protein homologous to tyrosine kinases. Mol Cell Biol. 1987; 7: 1378-85.
- Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993; 363: 458-60.
- Hofstra RMW, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994; 367: 375-6.
- Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993; 2: 851-6.
- Horibata S, Rice EJ, Mukai C, Marks BA, Sams K, Zheng H, et al. ER-positive breast cancer cells are poised for RET-mediated endocrine resistance. PLOS ONE. 2018; 13: e0194023-e0194043.
- Mulligan LM. GDNF and the RET receptor in cancer: new insights and therapeutic potential. Front Physiol. 2018; 9: 1873.
- Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005; 16: 441-67.
- Goodman KM, Kjær S, Beuron F, Knowles PP, Nawrotek A, Burns EM, et al. RET recognition of GDNF-GFRa1 ligand by a composite binding site promotes membrane-proximal self-association. Cell Rep. 2014; 8: 1894-904.
- Worby CA, Vega QC, Chao HH-J, Seasholtz AF, Thompson RC, Dixon JE. Identification and characterization of GFRa-3, a novel co-receptor belonging to the glial cell line-derived neurotrophic receptor family. J Biol Chem. 1998; 273: 3502-8.
- Amoresano A, Incoronato M, Monti G, Pucci P, de Franciscis V, Cerchia L. Direct interactions among Ret, GDNF and GFRa1 molecules reveal new insights into the assembly of a functional three-protein complex. Cell Signal. 2005; 17: 717-27.
- Wang X. Structural studies of GDNF family ligands with their receptors-insights into ligand recognition and activation of receptor tyrosine kinase RET. Biochim Biophys Acta. 2013; 1834: 2205-12.
- Mahato AK, Sidorova YA. RET receptor tyrosine kinase: role in neurodegeneration, obesity, and cancer. Int J Mol Sci. 2020; 21: 7108-28.
- Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994; 367: 380-3.
- Shakya R, Watanabe T, Costantini F. The role of GDNF/Ret signaling in ureteric bud cell fate and branching morphogenesis. Dev Cell. 2005; 8: 65-74.
- Pachnis V, Mankoo B, Costantini F. Expression of the c-RET proto-oncogene during mouse embryogenesis. Development. 1993; 119: 1005-17.
- Gould TW, Yonemura S, Oppenheim RW, Ohmori S, Enomoto H. The neurotrophic effects of glial cell line-derived neurotrophic factor on spinal motoneurons are restricted to fusimotor subtypes. J Neurosci. 2008; 28: 2131-46.
- Kramer ER, Knott L, Su F, Dessaud E, Krull CE, Helmbacher F, et al. Cooperation between GDNF/Ret and ephri-nA/EphA4 signals for motor-axon pathway selection in the limb. Neuron. 2006; 50: 35-47.
- Jain S, Naughton CK, Yang M, Strickland A, Vij K, Encinas M, et al. Mice expressing a dominant-negative Ret mutation phenocopy human Hirschsprung disease and delineate a direct role of Ret in spermatogenesis. Development. 2004; 131: 5503-13.
- Fonseca-Pereira D, Arroz-Madeira S, Rodrigues-Campos M, Barbosa IAM, Domingues RG, Bento T, et al. The neurotrophic factor receptor RET drives haematopoietic stem cell survival and function. Nature. 2014; 514: 98-101.
- Subbiah V, Cote GJ. Advances in targeting RET-dependent cancers. Cancer Discov. 2020; 10: 498-505.
- Wang H, Li Q, Zhang Z, Xiao P, Li L, Jiang Q. Functional studies on novel RET mutations and their implications for genetic counseling for Hirschsprung disease. Front Genet. 2019; 10: 924.
- Castinetti F, Moley J, Mulligan L, Waguespack SG. A comprehensive review on MEN2B. Endocr Relat Cancer. 2018; 25: T29-39.
- Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adeno-carcinoma. Nat Med. 2012; 18: 375-7.
- Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol. 2016; 12: 192-202.
- Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012; 18: 378-81.
- Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012; 22: 436-45.
- Qian Y, Chai S, Liang Z, Wang Y, Zhou Y, Xu X, et al. KIF5B-RET fusion kinase promotes cell growth by multilevel activation of STAT3 in lung cancer. Mol Cancer. 2014; 13: 176.
- Richardson DS, Gujral TS, Peng S, Asa SL, Mulligan LM. Transcript level modulates the inherent oncogenicity of RET/PTC oncoproteins. Cancer Res. 2009; 69: 486-4869.
- Hyndman BD, Crupi MJF, Peng S, Bone LN, Rekab AN, Lian EY, et al. Differential recruitment of E3-ubiquitin ligase complexes regulates RET isoform internalization. J Cell Sci. 2017; 130: 3282-96.
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448: 561-6.
- Suzuki M, Makinoshima H, Matsumoto S, Suzuki A, Mimaki S, Matsushima K, et al. Identification of a lung adenocarcinoma cell line with CCDC6-RET fusion gene and the effect of RET inhibitors in vitro and in vivo. Cancer Sci. 2013; 104: 896-903.
- Matsubara D, Kanai Y, Ishikawa S, Ohara S, Yoshimoto T, Sakatani T, et al. Identification of CCDC6-RET fusion in the human lung adenocarcinoma cell line, LC-2/ad. J Thorac Oncol. 2012; 7: 1872-1876.
- Huang Q, Schneeberger VE, Luetteke N, Jin C, Afzal R, Budzevich MM, et al. Preclinical modeling of KIF5B-RET fusion lung adenocarcinoma. Mol Cancer Ther. 2016; 15: 2521-9.
- Mukhopadhyay S, Pennell NA, Ali SM, Ross JS, Ma PC, Velcheti V. RET-rearranged lung adenocarcinomas with lymphangitic spread, psammoma bodies, and clinical responses to cabozantinib. J Thorac Oncol. 2014; 9: 1714-9.
- Chau NG, Haddad RI. Vandetanib for the treatment of medullary thyroid cancer. Clin Cancer Res. 2013; 19: 524-9.
- Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013; 3: 630-5.
- Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2016; 17: 984-93.
- Markowitz JN, Fancher KM. Cabozantinib: a multi-targeted oral tyrosine kinase inhibitor. Breast Cancer Res Treat. 2016; 160: 305-12.
- Houvras Y, Wirth LJ. Cabozantinib in medullary thyroid carcinoma: time to focus the spotlight on this rare disease. J Clin Oncol. 2011; 29: 2616-8.
- Song M. Progress in discovery of KIF5B-RET kinase inhibitors for the treatment of non-small-cell lung cancer. J Med Chem. 2015; 58: 3672-81.
- Wang M, Naganna N, Sintim HO. Identification of nicotinamide aminonaphthyridine compounds as potent RET kinase inhibitors and antitumor activities against RET rearranged lung adenocarcinoma. Bioorg Chem. 2019; 90: 103052.
- Heymach JV, Johnson BE, Prager D, Csada E, Roubec J, Pešek M, et al. Randomized placebo-controlled phase II study of vandetanib plus docetaxel in previously treated non-small-cell lung cancer. J Clin Oncol. 2007; 25: 4270-7.
- Heymach JV, Paz-Ares L, De Braud FD, Sebastian M, Stewart DJ, Eberhardt WEE, et al. Randomized phase II study of vandetanib alone or with paclitaxel and carboplatin as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2008; 26: 5407-15.
- Herbst RS, Sun Y, Eberhardt WEE, Germonpré P, Saijo N, Zhou C, et al. Vandetanib plus docetaxel versus docetaxel as secondline treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised. Phase 3 trial. Lancet Oncol. 2010; 11: 619-626.
- Wang M-C, Yang J-L, Gao T-M. Vandetanib plus docetaxel versus docetaxel for advanced non-small cell lung cancer: a meta-analysis. Chin J Evid Based Med. 2011; 11: 1151-5.
- Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth fac-tor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008; 14: 5459-65.
- Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008; 122: 664-71.
- Okamoto K, Kodama K, Takase K, Sugi NH, Yamamoto Y, Iwata M, et al. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion-driven tumor models. Cancer Lett. 2013; 340: 97-103.
- Tohyama O, Matsui J, Kodama K, Hata-Sugi N, Kimura T, Okamoto K, et al. Antitu-mor activity of lenvatinib. J Thyroid Res. 2014; 2014: 638747.
- Velcheti V, Hida T, Reckamp KL, Yang JC, Nokihara H, Sachdev P, et al. Phase 2 study of lenvatinib (LN) in patients (Pts) with RET fusion-positive adenocarcinoma of the lung. Ann Oncol. 2016; 27.
- Horiike A, Takeuchi K, Uenami T, Kawano Y, Tanimoto A, Kaburaki K, et al. Sorafenib treatment for patients with RET fusion-positive non-small cell lung cancer. Lung Cancer. 2016; 93: 43-6.
- Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, et al. Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N Engl J Med. 2020; 383: 813-24.
- Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas M,E et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018; 29: 1869-76.
- Markham A. Selpercatinib: first approval. Drugs. 2020; 80: 1865-70.
- Bradford D, Larkins E, Mushti SL, Rodriguez L, Skinner AM, Helms WS, et al. FDA approval summary: selpercatinib for the treatment of lung and thyroid cancers with RET gene mutations or fusions. Clin Cancer Res. 2021; 27: 2130-5.
- Kim J, Bradford D, Larkins E, Pai-Scherf LH, Chatterjee S, Mishra-Kalyani PS, et al. FDA Approval Summary: Pralsetinib for the treatment of lung and thyroid cancers with ret gene mutations or fusions. Clin Cancer Res. 2021; 27: 5452-6.
- Subbiah V, Shen T, Terzyan SS, Liu X, Hu X, Patel KP, et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper ret mutations. Ann Oncol. 2021; 32: 261-8.
- Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 2018; 8: 836-49.
- Gainor JF, Curigliano G, Kim D-W, Lee DH, Besse B, Baik CS, et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (AR-ROW): a multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021; 22: 959-69.
- FDA approves selpercatinib; pralsetinib may soon follow. Cancer Discov. 2020; 10: OF1.
- Wright KM. FDA approves pralsetinib for treatment of adults with metastatic RET fusion-positive NSCLC. Oncology (Williston Park). 2020; 34: 406.
- Sun F, McCoach CE. Therapeutic advances in the management of patients with advanced RET fusion-positive non-small cell lung cancer. Curr Treat Options Oncol. 2021; 22: 72.
- Horvath L, Pircher A, ASCO. ASCO 2020 non-small lung cancer (NSCLC) personal Highlights. Memo. 2021; 14: 66-9.
- Fu XY, Dong XD, Zeng L, Ashby Jr CR, Chen ZS, Cheng C. Pralsetinib for the treatment of non-small cell lung cancer. Drugs Today (Barc). 2021; 57: 559-69.
- Gainor JF, Lee DH, Curigliano G, Doebele RC, Kim D-W, Baik CS, et al. Clinical activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients (Pts) with advanced RET-fusion+ non-small cell lung cancer (NSCLC). J Clin Oncol. 2019; 37: 9008.
- Schoffski P, Cho BC, Italiano A, Loong HHF, Massard C, Medina Rodriguez LM, et al. BOS172738, a highly potent and selective RET inhibitor, for the treatment of RET-altered tumors including RET-fusion+ NSCLC and RET-mutant MTC: phase 1 study results. J Clin Oncol. 2021; 39: 3008.
- Schoffski P, Aftimos PG, Massard C, Italiano A, Jungels C, Andreas K, et al. A phase I study of BOS172738 in patients with advanced solid tumors with RET gene alterations including non-small cell lung cancer and medullary thyroid cancer. J Clin Oncol. 2019; 37: TPS3162.
- Schenck Eidam H, Russell J, Raha K, DeMartino M, Qin D, Guan HA, et al. Discovery of a first-in-class gut-restricted RET kinase inhibitor as a clinical candidate for the treatment of IBS. ACS Med Chem Lett. 2018; 9: 623-8.
- Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan AC, Yoda S, et al. Me-chanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann Oncol. 2020; 31: 1725-33.
- Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of ac-quired resistance to osimertinib in EGFR-mutant NSCLC and Clinical validation of combined EGFR and RET inhibition with osimertinib and BlU-667 for acquired RRT fusion. Cancer Discov. 2018; 8: 1529-39.
- Suda K, Mitsudomi T. Emerging oncogenic fusions other than ALK, ROS1, RET, and NTRK in NSCLC and the role of fusions as resistance mechanisms to targeted therapy. Transl Lung Cancer Res. 2020; 9: 2618-28.
- Larocque E, Naganna N, Ma X, Opoku-Temeng C, Carter-Cooper B, Chopra G, et al. Aminoi-soquinoline benzamides, FLT3 and Src-family kinase inhibitors, potently inhibit proliferation of acute myeloid leukemia cell lines. Future Med Chem. 2017; 9: 1213-25.
- Parate S, Kumar V, Hong JC, Lee KW. Putative dual inhibitors of mTOR and RET kinase from natural products: pharmacophore-based hierarchical virtual screening. J Mol Liq. 2022; 350: 118562-74.
- Solomon BJ, Tan L, Lin JJ, Wong SQ, Hollizeck S, Ebata K, et al. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J Thorac Oncol. 2020; 15: 541-9.
- Zhang L, Moccia M, Briggs DC, Bharate JB, Lakkaniga NR, Knowles P, et al. Discovery of N-trisubstituted pyrimidine derivatives as type I RET and RET gatekeeper mutant inhibitors with a novel kinase binding pose. J Med Chem. 2022; 65: 1536-51.
- Moccia M, Yang D, Lakkaniga NR, Frett B, McConnell N, Zhang L, et al. Targeted activity of the small molecule kinase inhibitor Pz-1 towards RET and TRK kinases. Sci Rep. 2021; 11: 16103.
- Drilon A, Rogers E, Zhai D, Deng W, Zhang X, Lee D, et al. TPX-0046 is a novel and potent RET/SRC inhibitor for RET-driven cancers. Ann Oncol. 2019; 30: v190-1.
- Fancelli S, Caliman E, Mazzoni F, Brugia M, Castiglione F, Voltolini L, et al. Chasing the target: new phenomena of resistance to novel selective RET inhibitors in lung cancer. Updated Evidence and Future Perspectives. Cancers. 2021; 13: 1091-113.
- Kolakowski GR, Anderson ED, Ballard JA, Brandhuber BJ, Condroski KR, Gomez EB, et al. Preclinical characterization of potent and selective next-generation RET inhibitors. Cancer Res. 2021; 81: 1464-1464.
- Miyazaki IS, Tadashi M. Kato, Hidenori Fujita, condensed pyrimidine compound or salt thereof. Japan: Applicant, Taiho Pharmaceutical Co. Ltd.; 2017. WO 201704355A1.
- Subbiah V, Zhong J, Lu Y, Liu Y, Chen M, Chen X, et al. The development of APS03118, a potent next-generation RET inhibitor for treating RET-inhibitor-resistant patients. J Clin Oncol. 2022; 40: e15107.
- Rebuzzi SE, Zullo L, Rossi G, Grassi M, Murianni V, Tagliamento M, et al. Novel emerging molecular targets in non-small cell lung cancer. Int J Mol Sci. 2021; 22: 2625-49.
- Tan L, Solomon BJ. Defining resistance mechanisms to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small cell lung cancer. Ann Oncol. 2020; 31: 1599-600.
- Zhang Y, Chan S, He R, Liu Y, Song X, Tu ZC, et al. 1-methyl-3-((4-(quinolin-4-yloxy)phenyl)amino)-1H-pyrazole-4-carboxamide derivatives as new rearranged during transfection (RET) kinase inhibitors capable of suppressing resistant mutants in solvent-front regions. Eur J Med Chem. 2022; 244: 114862.
- Román-Gil MS, Pozas J, Rosero-Rodríguez D, Chamorro-Pérez J, Ruiz-Granados Á, Caracuel IR, et al. Resistance to RET targeted therapy in thyroid cancer: molecular basis and overcoming strategies. Cancer Treat Rev. 2022; 105: 102372.
- Liu X, Shen T, Mooers BHM, Hilberg F, Wu J. Drug resistance profiles of mutations in the RET kinase domain. Br J Pharmacol. 2018; 175: 3504-15.
- Nakaoku T, Kohno T, Araki M, Niho S, Chauhan R, Knowles PP, et al. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat Commun. 2018; 9: 625.
- Yoda S, Lin JJ, Lawrence MS, Burke BJ, Friboulet L, Langenbucher A, et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 2018; 8: 714-29.
- Subbiah V, Shen T, Tetzlaff M, Weissferdt A, Byers LA, Cascone T, et al. Patient-driven discovery and post-clinical validation of NTRK3 fusion as an acquired resistance mechanism to selpercatinib in RET fusion-positive lung cancer. Ann Oncol. 2021; 32: 817-9.
- Rosen EY, Johnson ML, Clifford SE, Somwar R, Kherani JF, Son J, et al. Overcoming MET-dependent resistance to selective RET inhibition in patients with RET fusion-positive lung cancer by combining selpercatinib with crizotinib. Clin Cancer Res. 2021; 27: 34-42.
- Shen T, Hu X, Liu X, Subbiah V, Mooers BHM, Wu J. The L730V/I RET roof mutations display different activities toward pralsetinib and selpercatinib. npj Precis Oncol. 2021; 5: 48.