
Review Article
Austin J Pharmacol Ther. 2014; 2 (9).1048
PARP Enzymes are Potential Targets for Cancer Chemotherapy
Sabry M Attia* and Khalid A Al-Enazi
Department of Pharmacology and Toxicology, King Saud University, Saudi Arabia
*Corresponding author: : Sabry M Attia, Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, P.O. 2457, Riyadh 11451, Saudi Arabia
Received: August 28,, 2014; Accepted: September 25, 2014; Published: September 25, 2014
Abstract
PARP is a family of enzymes involved in a number of cellular processes involving mainly DNA repair and programmed cell death. PARP1 is a highly abundant nuclear enzyme that is constitutively expressed in response to DNA damage. Its main functions are to detect SSBs and to recruit the enzymatic machinery involved in DNA damage repair. Once PARP1 detects a SSB, it binds to the lesion and undergoes a structural change; it then initiates the synthesis of a PAR chain from NAD+. This synthesis signals for the other DNA repairing enzymes such as DNA ligase III, DNA polymerase beta and XRCC1. When DNA is mildly damaged, PARP1 is activated and participates in the DNA repair process, resulting in cell survival. However, in the case of extensive DNA damage, PARP1 is overactivated and rapidly depletes the intracellular NAD+ and ATP pools, which slows the rate of glycolysis and mitochondrial respiration, leading to cell dysfunction or even necrotic cell death. Thus, PARP1 overactivation has been shown to be involved in the pathogenesis of several diseases. Due to the dual involvement of PARP1 in the DNA damage response and cell death, the pharmacological modulation of PARP1 activity using PARP1 inhibitors constitutes a useful tool to increase the activity of DNA-binding anticancer agents. Moreover, PARP1 inhibitors may also be useful to restore cellular functions in many diseases and pathophysiological states. Thus, PARP1 may be considered a potential target for pharmacological intervention against various pathophysiological states. In this review, recent developments in our understanding of PARP1 inhibitors and its role in cancer and other diseases will be presented and discussed.
Keywords: PARP inhibitors; Pathophysiology; Cancer; Cancer therapy
Abbreviations
PARP: Poly (ADP-ribose) Polymerase; SSBs: Single-Strand Breaks; ADP: Adenosine Diphosphate; NAD+: Nicotinamide Adenine Dinucleotide; PAR: Poly (ADP) Ribose Polymer; MAR: Mono (ADP) Ribose Polymer; PARG: Poly(ADPR) Glycohydrolase; XRCC1: X-ray Repair Cross-Complementing 1; ATP: Adenosine Triphosphate
Introduction
PARP enzymes
Poly (ADP-ribose) polymerase-1 (PARP1) is a member of the PARP enzyme family consisting of PARP1 and several recently identified novel poly (ADP-ribosylating) enzymes. PARP-1 is an abundant nuclear protein functioning as a DNA nick-sensor enzyme. Upon binding to DNA breaks, activated PARP cleaves NAD(+) into nicotinamide and ADP-ribose and polymerizes the latter onto nuclear acceptor proteins including histones, transcription factors, and PARP itself [1]. The addition of negatively charged polymers profoundly alters the properties and functions of the target proteins. Poly (ADP-ribosyl) action is involved in the regulation of many cellular processes such as DNA repair, gene transcription, cell cycle progression, cell death, chromatin functions and genomic stability [2]. Besides the 7 members of the PARP family characterized so far, additional 11 molecules that may have potential PARP activity, have been fished out from the analysis of protein database [3]. In fact, all these proteins share the so-called “PARP signature”, which is located within the catalytic site [4].
In regard to DNA damage signaling, the PARP family of enzymes catalyses a post-translational protein modification reaction that can occur immediately after exposure of cells to DNA damaging agents [5]. In vivo, 90% of PAR deriving by the use of β-NAD+ substrate, are attached to the auto-modification domain of PARP1, the main enzyme catalyzing this reaction. Furthermore, PARP1 has been found to covalently poly (ADP-ribosyl) ate a number of nuclear components (hetero-modification) including both structural and functional proteins [5]. Moreover, PAR chains (up to 200 residues long) linked to PARP1 are able to interact with several target proteins containing a ‘‘polymer binding motif’’ [6] thereby modulating their function; for instance, p53 specific binding to its DNA consensus sequence can be regulated by such a mechanism. Poly (ADPR) Glycohydrolase (PARG) is the PARPs’ enzymatic counterpart catalysing PAR catabolism [5].
The role of PARP1 in facilitating DNA repair of both single and double strand breaks has been clearly demonstrated, due to its involvement in the base excision repair and non-homologous end joining pathways, respectively. In fact, PARP1 binds to DNA strand breaks formed by ionizing radiation or through the incision of apurinic/apyrimidinic sites during the repair of the damage induced by anticancer agents by means of base excision repair [7]. PARP-2 interacts with PARP1 and shares common partners involved in the single strand break repair and base excision repair pathways: X-Ray Repair Cross-Complementing 1 (XRCC1), DNA polymerase β and ligase III [8]. Both enzymes detect DNA strand interruptions acting as nick sensors, providing rapid signals to halt transcription and recruiting the enzymes required for DNA repair at the site of damage. Therefore, PARP1 is critical for the maintenance of genomic stability through the regulation of DNA repair mechanisms.
Interestingly, PARP1 deficiency does not seem to be such a problem for non-malignant cells. As a matter of fact, mice engineered to lack PARP1 are both viable and fertile [9]. These PARP1 mice do not seem to develop early onset tumours. However, conflicting results have been reported in another paper suggesting that PARP genetic ablation may predispose to mammary cancer [10]. The explanation for this apparent lack of ‘severe consequences’ as a result of PARP deficiency can be explained by an understanding of DNA repair mechanisms. The loss of PARP1 activity affects repair of single-strand breaks via base excision repair, resulting in cells using different DNA repair pathways. At the time of DNA replication, unrepaired single-strand breaks are converted into double-strand breaks at replication forks and repaired by homologous recombination. PARP1 inhibition blocks repair of single-strand breaks but repair of double-strand breaks is able to proceed [11]. In this circumstance, homologous recombination activity is increased and acts as a very efficient error-free rescue mechanism.
PARP inhibitors and cancer chemotherapy
The particular behavior of PARP1has made it a primary cellular target for a number of novel compounds considerably more effective to cells that contain high levels of PARP1and which are undergoing high rates of DNA damage. As early as in 1980, Durkacz and colleagues used the still immature, low-potency PARP inhibitor 3-aminobenzamide to derail DNA damage repair and enhance the cytotoxicity of the DNA alkylating agent dimethyl sulfate [12]. Since then, a huge number of preclinical studies legitimated PARP inhibitors as sensitizing agents to DNA-damaging drugs and radiotherapy [13,14]. In regard to chemopotentiation, the data obtained with chemical inhibitors of PARP or derived from PARP1 or PARP-2 knock-out mice clearly indicate that suppression of PARP activity increases cell susceptibility to DNA damaging agents and inhibits strand break rejoining [15].
The vast majorities of PARP inhibitors developed to date interact with the nicotinamide binding domain of the enzyme and behave as competitive inhibitors with respect to NAD+. Structural analogues of nicotinamide, such as benzamide and derivatives were among the first compounds to be investigated as PARP inhibitors. However, these molecules have a weak inhibitory activity and possess other effects unrelated to PARP inhibition. Using the rational drug design approach, more potent inhibitors of the enzyme have been developed, which are selective for PARP inhibition. In vitro and in vivo analyses revealed that these compounds were able to potentiate the efficacy of chemotherapeutic agents using both human and murine tumor models [15-17] and synergistic chemosensitivity of various breast and other cancer cell lines to PARP inhibition and cisplatin or other antineoplastic drugs has been reported [18].
In several clinical trials inhibition of PARP alone or in combination with DNA-damaging anticancer agents showed considerable promise in facilitating tumor cell death [19]. In an exciting finding, PARP1 has recently been implicated in the chemoresistance of cancer cells to cisplatin, which is a significant clinical problem [20] and PARP inhibition results in a synergistic chemosensitivity of triple-negative breast cancer cell lines to gemcitabine and cisplatin [21]. Furthermore, several PARP inhibitors (alone or in combination with other antineoplastic agents) from Pfizer (AG014699/PF-01367338), AstraZeneca (AZD2281/KU-0059436/olaparib), Sanofi-Aventis (BSI-201/SAR240550/iniparib), Abbott Laboratories (ABT-888/ veliparib), Merck (MK4827), and Cephalon (CEP-9722) are in clinical development for various forms of primary breast, ovarian, brain, pancreatic, peritoneal, intestinal, and colorectal cancer; leukemia; lymphoma; and neoplasm metastasis [22].
Several of these recent clinical trials are aiming to evaluate the efficacy of several PARP inhibitors (AVT-888, PF-01367338, BSI-201, AZD2281) in combination with the platinum compound carboplatin to treat ovarian, fallopian tube, primary peritoneal and triple-negative primary or metastatic breast cancer [22]. PARP inhibitors appear to be more active in vivo as enhancers of the antitumor activity of cisplatin than they are in cultured cells. For example, CEP-6800 and BGP-15 failed to enhance cisplatin cytotoxicity in a variety of cell lines in vitro, but did enhance cisplatin-induced reduction in the growth of several xenograft models [23,24]. ABT-888 also enhanced the antitumor activity of cisplatin against BRCA mutant human breast cancer xenografts without any cell based data being described [25]. Similarly, there is xenograft evidence of the enhancement of doxorubicin and cyclophosphamide that is not supported by cell-based studies [25].
The mechanisms of the beneficial effects of PARP inhibitors in cancer are multiple and may involve attenuation of cancer cell proliferation and migration, promotion of cancer cell demise, decrease of angiogenesis, and modulation of the tumor environment (e.g., attenuation of inflammation). The selective promotion of cell death in cancer, but not in normal cells, by PARP inhibitors is based on the novel approach of “synthetic lethality” in cancer therapy. The rationale for this is that in cancers with selective defects in homologous recombination repair (cancer cells frequently harbor defects in DNA repair pathways leading to genomic instability), inactivation of PARPs directly causes cell death [14].
Besides being involved in DNA repair, PARP may also act as a mediator of cell death (Figure 1). In fact, extensive DNA damage, that saturates cell repair ability, is known to trigger PARP overactivation with consequent extensive NAD consumption during the synthesis of ADP-polymers, which leads to ATP depletion, slowing the rate of glycolysis, electron transport, and ATP formation, eventually leading to induction of mutagenesis or death of various normal cell types [26,27]. Extensive PARP activation may also results in caspase-independent programmed cell death, mediated by the translocation of apoptosis inducing factor to the nucleus [28]. Mutagenesis may lead to the development of secondary tumours, while necrosis will triggers inflammation and tissue injury that may lead to DNA damage again. Moreover, in extensive DNA damage, the inhibition of PARP activity will inhibits NAD+ and ATP depletion, thus allowing cells to function normally, or, if the apoptotic process has initiated the cell will die by apoptosis instead of necrosis.
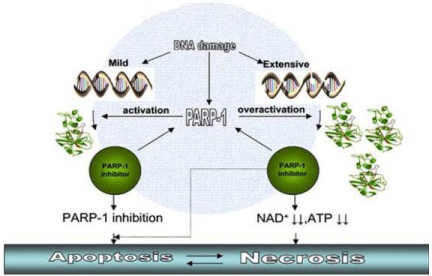
Figure 1: PARP-1 enzyme acts as a mediator of cell death. Activation of PARP-1 may induced by mild DNA damage. the use of PARP-1 inhibitors may result in efficient block of DNA repair and subsequent apoptotic cell death. Overactivation of PARP-1 may induced by very intense DNA damage and subsequent so that depletion of NAD+ and ATP stores to lethal-inducing levels may lead to necrotic tumor cell death. In this scenario, the use of PARP-1 inhibitors may block PARP-1 overactivation and, then, cell death is switched from necrosis to apoptosis [13].
PARP inhibitors to decrease toxicity induced by cancer chemotherapy
Another application of PARP inhibitors regards their use in combination with chemotherapy to reduce drug-induced cardiactoxicity by avoiding necrotic cell death. The clinical use of doxorubicin is limited by its serious dose-dependent cardiotoxicity, which leads to irreversible degenerative cardiomyopathy and heart failure. It has been demonstrated that the impairment of cardiac function in doxorubicin-induced heart failure is due to oxidative stress. Increased formation of ONOO- and H2O2 causes single strand breaks that in turn trigger PARP activation within the heart. The PARP inhibitor PJ34 exerts protective effects against cardiacdys function and PARP-1-/- animals are more resistant to the cardiotoxicity of the chemotherapy with respect to their PARP- 1+/+ counterparts [29]. However, PARP inhibition does not prevent metalloproteinase activation, which is dependent on oxidative stress and plays an important role incongestive heart failure and reperfusion injury. This suggests that PARP activation occurs downstream from the generation of oxidant species.
The nicotinic amidoxime derivative (BGP-15), a novel PARP inhibitor has been shown to protect animals against nephrotoxicity of cisplatin without affecting its antitumor efficacy [30]. Cisplatin-induced nephrotoxicity is a serious adverse effect that is characterized by reduced renal blood flow and proximal tubule injury. Prevention of kidney damage generally relies on intensive intravenous hydrationin combination with forced diuresis. Moreover, various free radical scavengers such as compounds containing thiolgroups such as amifostine, or sodium thiosulfate have shown some protective effect. However, no treatment provides complete protection especially against long-term damage and nephrotoxicity is still a major limit of cancer chemotherapy with cisplatin. Although the exact mechanism of cisplatin-induced nephrotoxicity is not yet clear, activation of PARP has been implicated in kidney tubular injury. It is known that cisplatin generates reactive oxygen species, which in turn cause DNA single strand breaks and PARP activation. In fact, poly (ADP-ribosyl) ation followed by ATP depletion has been demonstrated in renal tubular cells exposed to cisplatin and both effects were reversed by the PARP inhibitor 3-aminobenzamide [31]. Interestingly, BGP-15 has also a protective effect against peripheral neuropathy associated with cisplatin or with the antimicrotubule agent taxol [32]. In this case, it has been speculated that the neuroprotective effect of the PARP inhibitor is based on the prevention of free radical damage that causes the changes ofnerve conduction velocity induced by both drugs.
Potential applications of PARP in other diseases
One of the major and most promising research areas of PARP inhibition is the treatment of cancers. But there is emerging evidence that inhibition of the cellular poly(ADP-ribosyl)ation system may also have positive effects in the therapy of other diseases, as was shown in a variety of cell culture systems as well as animal models of ischemia/reperfusion injury, lung inflammation, shock, diabetes, and diabetic complications, among many others [2,14,18,33-35]. Evidence is emerging that the inhibition of PARPs activity can have beneficial effects for the organism under certain conditions, highlighting the double-faced function of PARPs. Treatment of tumor cells with a genotoxic chemotherapeutic agent and simultaneous inhibition of poly (ADP-ribosyl)ation enhances the apoptotic cell-killing effect of the agent by suppressing base-excision repair; on the other hand, the deleterious consequences of tissue damage by ischaemia-reperfusion and inflammation can be suppressed by inhibition of polymer formation, by blocking the necrotic pathway and/or by interfering with NF-κB signaling. It has been suggested that the positive effects of PARP inhibitors are related to the modulation of PARP1 activity, the protection of mitochondria from oxidative damage or the inhibition of the metabolic activation of the toxic compound [36,37].
Conclusions
From the above reported data, the advantage of the use of PARP inhibitors in cancer therapy is two-fold: to enhance the efficacy of DNA damaging anticancer drugs or, in selected cases, to protect normal tissues from the toxic effects of chemotherapy. Although apparently contradictories, these two indications clearly depend on the type of DNA damage provoked by the anticancer agent to be combined with PARP inhibitor. The first indication applies to chemotherapeutic agents that directly induce genotoxic damage, which requires PARP for its repair. On the other hand, the use of PARP inhibitors to counteract the untoward effects of chemotherapy concerns anticancer agents that provoke cell death generating oxidative stress and consequent PARP overactivation.
Acknowledgement
This work was supported by National Plan for Science and Technology (NPST), grant number 12-MED2648-02 in the Kingdom of Saudi Arabia.
References
- Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun. 2014; 5: 4426.
- Jagtap P, Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005; 4: 421-440.
- Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004; 26: 882-893.
- Weaver AN, Yang ES. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front Oncol. 2013; 3: 290.
- Bürkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+. FEBS J. 2005; 272: 4576-4589.
- Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem. 2000; 275: 40974-40980.
- Satoh MS, Poirier GG, Lindahl T. NAD(+)-dependent repair of damaged DNA by human cell extracts. J Biol Chem. 1993; 268: 5480-5487.
- Schreiber V, Amé JC, Dollé P, Schultz I, Rinaldi B, Fraulob V, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002; 277: 23028-23036.
- Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, et al. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995; 9: 509-520.
- Tong WM, Yang YG, Cao WH, Galendo D, Frappart L, Shen Y, et al. Poly(ADP-ribose) polymerase-1 plays a role in suppressing mammary tumourigenesis in mice. Oncogene. 2007; 26: 3857-3867.
- Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene. 2004; 23: 3872-3882.
- Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980; 283: 593-596.
- Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Soto M, et al. Poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors in cancer chemotherapy. Recent Pat Anticancer Drug Discov. 2006; 1: 39-53.
- Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med. 2013; 34: 1217-1256.
- Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, et al. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Cancer Res. 2000; 6: 2860-2867.
- Bowman KJ, White A, Golding BT, Griffin RJ, Curtin NJ. Potentiation of anti-cancer agent cytotoxicity by the potent poly(ADP-ribose) polymerase inhibitors NU1025 and NU1064. Br J Cancer. 1998; 78: 1269-1277.
- Clark CC, Weitzel JN, O'Connor TR. Enhancement of synthetic lethality via combinations of ABT-888, a PARP inhibitor, and carboplatin in vitro and in vivo using BRCA1 and BRCA2 isogenic models. Mol Cancer Ther. 2012; 11: 1948-1958.
- Mukhopadhyay P, Horváth B, Kechrid M, Tanchian G, Rajesh M, Naura AS, et al. Poly(ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic Biol Med. 2011; 51: 1774-1788.
- Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: Old and new paradigms revisited. Biochim Biophys Acta. 2014; 1846: 201-215.
- Ganesan S. MYC, PARP, and chemoresistance: BIN there, done that? Sci Signal. 2011; 4: pe15.
- Hastak K, Alli E, Ford JM. Synergistic chemosensitivity of triple-negative breast cancer cell lines to poly(ADP-Ribose) polymerase inhibition, gemcitabine, and cisplatin. Cancer Res. 2010; 70: 7970-7980.
- Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011; 10: 351-364.
- Racz I, Tory K, Gallyas F Jr, Berente Z, Osz E, Jaszlits L, et al. BGP-15 - a novel poly(ADP-ribose) polymerase inhibitor - protects against nephrotoxicity of cisplatin without compromising its antitumor activity. Biochem. Pharmacol. 2002; 63: 1099-1111.
- Miknyoczki SJ, Jones-Bolin S, Pritchard S, Hunter K, Zhao H, Wan W, et al. Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Can Ther. 2003; 2: 371-382.
- Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007; 13: 2728-2737.
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999; 96: 13978-13982.
- Tentori L, Portarena I, Graziani G. Potential clinical applications of poly(ADP-ribose) polymerase (PARP) inhibitors. Pharmacol Res. 2002; 45: 73-85.
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002; 297: 259-263.
- Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Haskó G, et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002; 300: 862-867.
- Racz I, Tory K, Gallyas F Jr, Berente Z, Osz E, Jaszlits L, et al. BGP-15 - a novel poly(ADP-ribose) polymerase inhibitor - protects against nephrotoxicity of cisplatin without compromising its antitumor activity. Biochem Pharmacol. 2002; 63: 1099-1111.
- Shino Y, Itoh Y, Kubota T, Yano T, Sendo T, Oishi R. Role of poly(ADP-ribose)polymerase in cisplatin-induced injury in LLC-PK1 cells. Free Radic Biol Med. 2003; 35: 966-977.
- Bárdos G, Móricz K, Jaszlits L, Rabloczky G, Tory K, Rácz I, et al. BGP-15, a hydroximic acid derivative, protects against cisplatin- or taxol-induced peripheral neuropathy in rats. Toxicol Appl Pharmacol. 2003; 190: 9-16.
- Ahmad SF, Zoheir KM, Bakheet SA, Ashour AE, Attia SM. Poly(ADP-ribose) polymerase-1 inhibitor modulates T regulatory and IL-17 cells in the prevention of adjuvant induced arthritis in mice model. Cytokine. 2014; 68: 76-85.
- Ahmad SF, Attia SM, Zoheir KM, Ashour AE, Bakheet SA. Attenuation of the progression of adjuvant-induced arthritis by 3-aminobenzamide treatment. Int Immunopharmacol. 2014; 19: 52-59.
- Tao R, Kim SH, Honbo N, Karliner JS, Alano CC. Minocycline protects cardiac myocytes against simulated ischemia–reperfusion injury by inhibiting poly(ADP-ribose) polymerase-1. J Cardiovasc Pharmacol. 2010; 56: 659-668.
- Szabados E, Literati-Nagy P, Farkas B, Sumegi B. BGP-15, a nicotinic amidoxime derivate protecting heart from ischemia reperfusion injury through modulation of poly(ADP-ribose) polymerase. Biochem Pharmacol. 2000; 59: 937-945.
- Cover C, Fickert P, Knight TR, Fuchsbichler A, Farhood A, Trauner M, et al. Pathophysiological role of poly(ADP-ribose) polymerase (PARP) activation during acetaminophen-induced liver cell necrosis in mice. Toxicol Sci. 2005; 84: 201-208.