
Review Article
Austin J Pharmacol Ther. 2015; 3(2).1070.
Basic Pharmacology of Bradykinin Receptor Agonists
Sharma JN* and Narayanan P
Department of Pharmacology and Therapeutics, Faculty of Pharmacy, Kuwait University, Health Sciences Centre, Kuwait
*Corresponding author: : Sharma JN, Department of Pharmacology and Therapeutics, Faculty of Pharmacy, Kuwait University, Health Sciences Centre, P O Box 24923, Safat 1311, Kuwait
Received: November 06, 2014; Accepted: June 18, 2015; Published: June 23, 2015
Abstract
Kinins are the most potent biologically active polypeptides, which are located in the vascular smooth muscle as well as in the heart. They produce their pharmacological effects by acting on G-protein-coupled constitutive kinin B2 or inducible kinin B1 receptors linked to signaling pathways involving increased intracellular Ca++ concentrations and release of nitric oxide (NO). Pharmacological studies on hypertension, cardiac failure, ischaemia, and myocardial infarction and left ventricular hypertrophy indicate that the reduced activity of the local kallikrein-kinin system (KKS) may be instrumental in the induction of cardiovascular disorders. The ability of kallikrein gene delivery and the use of kinin B2 receptor agonists to produce a wide spectrum of beneficial effects make it a powerful candidate in treating hypertension, cardiovascular and renal diseases. Strategies that activate kinin receptors might be applicable to the treatment of cardiovascular disease.
Keywords: Kinins; Kinin receptor agonists; Cardiac diseases; Cardiac protection
Introduction
Kinins are potent vasorelaxant polypeptides located in the vascular smooth muscle as well as in the heart. A number of observations obtained from clinical and experimental models of hypertension, cardiac failure, ischaemia, myocardial infarction and left ventricular hypertrophy, have suggested that KKS may be involved in the induction of cardiovascular-related diseases. Evidence suggests that the KKS may play a role in the central regulation of blood pressure in hypertensive rats [1]. Reduction in peripheral and cardiac KKS components may also be the cause of developing high blood pressure in both man and experimental animals. Kinins administered locally exert beneficial cardiac effects [2]. In vitro and in vivo studies suggested that BK reduce the duration and incidence of ischaemia in isolated rat hearts [3]. Studies undertaken in rats, dogs and humans revealed that kinins are released under conditions of ischaemia and myocardial infarction [4]. BK antagonists worsen ischaemic-induced effects 1BKs can contribute to the cardioprotective effects of preconditioning [1]. On the other hand, the reduction in cardiac infarct size by BK, after preconditioning in rabbits was prevented by a BK antagonist Icatibant (HEO 140) treatment [5]. Kinins have a role in protecting the heart against developing left ventricular hypertrophy by releasing nitric oxide [5]. It has been suggested that kinin B2 receptors agonists offer promising therapeutic approaches for the development of drugs for the treatment of cardiovascular diseases [6]. However, none of the currently known potent and selective peptide and nonpeptide agonists of kinin B2 receptor such as RMP-7, JMV-1116, FR- 190997 and FR-191413 have been selected for a clinical assessment in cardiovascular indications [6]. The objective of this review is to discuss the current concepts of BK antagonists.
The Kallikrein-Kinin System
The kinin family mainly includes BK (Arg-Pro-Pro-Gly-Phe-Ser- Pro-Phe-Arg), Kallidin (Lys-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe- Arg) and methionyl-lysyl-BK (Met-Lys-Arg-Pro-Pro-Gly-phe-Arg) [1,5,7]. These are pharmacologically active polypeptides derived from circulating precursors (kininogens) by the action of serine proteases, called kallikreins [1,5,7]. Once released into the circulation, kinins are rapidly (<15 sec) inactivated by enzymes called kininases [1,5,7].
Kininogens are typical secretory multifunctional proteins derived from a2-globulin [1,5,7]. These are synthesized in the liver and circulate in the plasma and body fluids. Two forms of kininogens are present in mammalian circulation, High Molecular Weight Kininogen (HMWK) and Low Molecular Weight Kininogen (LMWK) [1,5,7]. However, they have completely different biochemical, immunologic and functional characteristics [1,2]. In addition, there is a T-kininogen in the rat myocardium, which is considered to be an acute phase reactant of inflammation [8]. This kininogen releases T-kinin by the enzymatic action of T-kallikrein in rats [8].
Tissue kallikrein is found more widely distributed in the tissues (organs), such as kidney (urine), pancreas, salivary glands, intestine, prostate gland and synovial tissue [1,5,7]. These kallikreins are single chain acidic glycoproteins that differ from one another in molecular weight, biological function, and physicochemical and immunological properties [1,5,7]. In tissues the inactive enzyme, tissue kallikrein, is converted into the active form by the cleavage of an amino terminal peptide. The active tissue kallikrein liberates, Kallidin from LMWK [1,5-7]. Plasma kallikrein circulates in an inactive state also known as the pre-kallikrein or Fletcher factor. Inactive prekallikrein can be activated to form kallikrein by activated Hageman factor or factor XIIa, which then liberate BK from the HMWK [1,5-7]. In addition, plasma kallikrein is able to convert inactive factor XII to XIIa by positive feedback reaction [1]. The plasma prekallikrein and HMWK are present together in a complex form [1,5]. Factor XIIa and factor XI circulate with HMWK in the bound form [1,5-7]. Inactive factor XI is converted to active factor XIa through HMWK to participate in the intrinsic coagulation pathway [1,5-7].
Kininases, kinin-inactivating enzymes, are present in the plasma, urine, tissue, endothelial cells and body fluids [1,5]. Their prime function is to monitor the required BK concentrations in the body to perform the necessary physiological activities [1,5-7]. Carboxypeptidases or kininases I cleave the C-terminal arginine from BK and kallidin to produce their corresponding des-Arg derivatives [6]. Two metalloproteinases, neutral endopeptidase (NEP) and angiotensin-converting enzyme (ACE) also called kininases II degrade BK and kallidin into inactive fragments [1,5-7,9]. ACE is regarded as the most important kinin-degrading enzyme in the cardiovascular system and kidney [5-7]. It has a higher affinity for BK than for angiotensin I (Ang I), resulting in more favourable kinetics for BK than for angiotensin degradation [6]. Hence, ACE that can be inhibited by ACE inhibitors pharmacologically links the KKS with the renin-angiotensin system (RAS). Both ACE- and NEP- inhibitors increase BK peptide levels [9]. ACE- and NEP- inhibitors have different effects on kinin peptide levels in blood, urine, and tissue that may account for the differential contribution of ACE and NEP to kinin peptide metabolism in the multiple compartments in which kinin peptide generation occurs [9]. Figure 1 shows the mode of kinin formation.
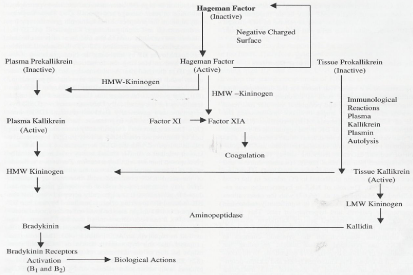
Figure 1: Bradykinin formation and its metabolism.
Kinin peptides are implicated in many physiological and pathological processes [7]. Some of the major physiological roles of kinins were described in parallel with their discovery [7]. Accumulating evidence suggests that kinins act as mediators of endogenous cardioprotective mechanisms [1-3,7]. Kinin had been shown to stimulate smooth muscle, produce hypotension and protect against ischaemia as will be discussed later in details. However, kinins also participate in the cardinal features of inflammation, producing swelling (edema), redness and heat (vasodilatoin) and pain [7]. This stimulated a large number of studies on the role of kinins in inflammation, tissue damage, antigen-antibody reactions and shock [7]. Moreover, BK-like activity was detected in the spinal fluid of individuals with degenerative diseases of the central nervous system, or in case of migraine and chronic schizophrenia [7]. Also, the bronchoconstriction action of intravenous BK has been documented [7]. Therefore, there is a need to understand how the generation of kinin peptides are regulated if we are to exploit the beneficial actions of these peptides and avoid their adverse effects.
The molecular basis of these contributions are through activation of B1 and / or B2 BK receptors and downstream activation of various second messengers system like nitric oxide (NO), cGMP, cAMP, arachidonic acid and inositol phosphates (IP3) that leads to increase in intracellular Ca+2 levels [1,6].
Kinin receptors classification
Kinins exert a variety of biological actions by acting through specific receptors that are widespread and belong to two major categories B1 and B2 that have been defined pharmacologically using a variety of peptidergic agonists and antagonists [10]. Recent pharmacologically findings from various studies suggest the existence of new receptor types, named B3, B4 and B5 [10]. These receptors mediate the contractile and the relaxant response of the opossum esophagus to BK [10]. However, B3, B4, and B5 receptors have not been sufficiently characterized either with agonists or with antagonists to be considered as new functional sites [10].
B1 and B2 bradykinin receptors
Regoli et al. in 2001 proposed the basic classification of kinin receptors into B1 and B2 receptors [10]. The B2 receptors normally predominate whereas B1 receptors are induced by tissue injury [7]. Further, and in contrast to B2 receptors, B1 receptors have been demonstrated only in certain species [1,5]. B2 receptors have been cloned and belong to the family of receptors with seven trans membrane-spanning domains and are G-protein couple to a number of biochemical pathways via a system of second messengers [1,5,7]. The B1 receptor displays high affinity and is preferentially activated by the kinin metabolites lacking the C-terminal arginine residue (des-Arg9-BK and des-Arg9-kallikdin) [1,5,7]. The B1 receptor is rarely expressed in certain vascular tissues, but it can be expressed in response to inflammation and tissue injury [1,5,6]. B1 receptor activation may induce stimulation of smooth muscle, increased cell proliferation, and collagen synthesis [1,5]. Agata et. al in 2000 evaluated the effect of KKS on the proliferation and migration of primary cultured vascular smooth muscle cells (VSMCs) in vitro, and neointima formation in balloon-injured rat carotid arteries in vivo. The results indicate that the B1 receptor contributes to the reduction of neointima formation via the promotion of reendothelialization [11]. B1 receptors were originally defined in terms of agonist rank order of potencies, des-Arg9-BK being the most potent relative to BK and this, along with an appreciable affinity for an antagonist, des- Arg9-[Leu8] -BK, was taken to demonstrate B1 receptors in various cells and tissues [1,5]. The majority of the prominent physiological actions of BK seem to be mediated by the stimulation of the constitutive BK B2 receptor [1,5,6]. B2 receptors exhibit higher affinity for BK and kallidin [5]. BK activates sensory nerve terminals, induces the release of pro-inflammatory and hyperalgesic mediators such as neuropeptidase, leukotrienes and cytokines, increases vascular permeability and induces vasodilatoin via stimulation of BK B2 receptors [6]. These pharmacological effects are the underlying cause of the strong pro-inflammatory and nociceptive properties of BK [6]. Therefore, B2 receptors may participate in pathological conditions such as septic shock, pancreatitis, edema, asthma, rhinitis, colitis, arthritis and pain [6]. These diseases might be promising target for the development of BK B2 receptor antagonists [6]. It has been shown that kinin acts on B2 receptors to release NO and prostaglandins (PGI2 and PGE2) [1,5,6]. These are the underlying cause of its vasodilatory, antihypertensive and antiatherosclerotic action [1,5,6]. Therefore, stimulation of BK B2 receptors might be a potential in the treatment and prevention of cardiovascular diseases such as hypertension, cardiac failure and ischaemia and myocardial hypertrophy [1,5,6]. Antagonist affinity has been pursued for more than two decades [1,5,12]. Des-Arg9-(leu8) -BK was the first introduced B1 receptor antagonist by Regoli [1,5,10,12]. The second category of antagonists came with the introduction of Icatibant (HOE 140), which shows high affinity in all preparations, has no residual agonistic activities and very selective B2 receptor antagonist [1,12]. The third category of antagonists, exemplified by FR-167344, FR-173657, and Bradyzide that are extremely potent, selective and orally active non-peptide BK B2 receptor antagonists [1,5,12]. These BK receptor antagonists will allow rapid progress in analyzing the pathological states that relate to hyperactivity of kinins [1,5,12]. Table 1 shows BKs receptor, functions and antagonists.
![]()
studies
applicatio
Ligands
- Rabbit
- rodent
- Metabolically stable
- High affinity and selectivity
- Hypertension
- Stimulation of vascular formation following ischemia
Agonists
R-838(Sar-[D-Phe8]des-Arg9-BK)
- Rat
- Mice
- Pain
- Ischemic vascular disease
Antagonists
[Leu8]des-Arg9-BK
- Human B1 receptor antagonist
- Optimal B1 receptor antagonist
Lys-[Leu8]des-Arg9-Bk
- Rabbit
- Metabolically stable (not very potent compared with the affinity of reference compound Lys-[Lue8]des-Arg9-BK
Ac-Lys-[MeAla6,Leu8]des-Arg9-BK
- Human and rabbit B1 receptor
- Mice
- High affinity
- Allergic lung inflammation
R-715(Ac-Lys-[�D-Nal7,Ile8]des-Arg9-BK
- Human and rabbit B1 receptor
- Fairly high selectivity for B1 receptor due to Lys
- Metabolically stable residue
B9858(Lys-Lys-[Hyp3,Igl5,D-Igl7,Oic8]des-Arg9-BK)
- Rabbit jugular vein, guinea pig ileum, rabbit aorta
- Residual antagonistic effects on B2 receptor
- Moderate affinity
Des-Arg10-HOE 140
- Demonstration of compatibility of B1 and B2 receptors structure by the accommodation of a single pharmacophore
- Mixed B1 and B2 receptor antagonist even if desArg9 fragment has substantial selectivity for B1 receptor
B9430 (D-Arg-[Hyp3,Igl5,D-Igl7,Oic8]-BK)
- Mice
- Rat model
- Allergic lung inflammation
- Air way allergy
R-954(Ac-Orn-[Oic2,a-MePhe5,D-�Nal7,Ile8]des-Arg9-BK)
- Human receptor (no in vivo data)
- Potent and competitive B1 receptor antagonist
- High affinity
PS020990
- Human and rat B1 receptor in vitro
- Rat and dog
- Rabbit aortic preparation, rabbit jugular vein
- Mice and rat
- Selective antagonist
- Powerful and selective antagonist
- Hyperalgesia
- Speculative on pain, inflammation and sepsis
- Inflammation and hyperalgesia
-Compound 12 (benzodiazepine based structure)
Benzo-sulfonylamide compounds
- Compound 12
- Compound 11
- SSR240612
- In vivo rodent models
- Human: phase II studies on glioma
- Vascular permeability (blood brain barrier): adjuvant to chemotherapy of brain tumors
Agonists
Labradimil ([Hyp3, Thi5, 4-MeTyr8 ψ(CH2-NH)Arg9]-BK)
- Rat
- Hypertension
FR190997
- Rat uterus, guinea pig ileum
- Rat uterus, guinea pig ileum
- low potency, antagonist/ partial agonist activity
- Potent antagonist, no agonist activity
Antagonists
First generation
[D-Phe7]-BK
[Thi5,8, D-Phe7]-BK
- Animal model (high affinity for the human, rabbit and guinea pig B2 receptor)
- Human, nasal treatment
- Human
- Rat
- High affinity, long lasting, competitive activity
- No residual agonist effect
- Resistance to peptidases
- Acute rhinitis
- Asthma
- Early stage of inflammation
- Persistent inflammatory pain
Second generation
HOE 140 (Icatibant; D-Arg-[Hyp3, Thi5, D-Tic7, Oic8]-BK)
- On human tissue
- For guinea pig B2 receptor
- In active
- Limited affinity
Third generation
Phosphonium family:
WIN64338
- Identification of the absolute requirement for B2 receptor binding affinity: presence of two positive charges at a distance about 10 A� separated by a lipophilic residue, playing the role of the Phe8 side chain in the native ligand
-Micromolar binding affinity to human B2 receptor
WIN62318
- Oral activity at doses ranging between 1 and 30 mg/kg in different tests and species.
- High B2 receptor affinity and selectivity versus B1 receptor
Quinoline and imidazole [1,2-a]pyridine family:
- Rat and mice
- Oral activity on hyperalgesia and inflammation
FR165649,
FR173657,
FR184280
- Guinea pig ileum, human A-431 cells
- Guinea pigs (oral activity) designed as clinical candidate to treat inflammatory disease
- Selective and high potent binding activity
- bronchoconstriction
FR167344
- Human B2 receptor
- High affinity
Compound 38
- Human B2 receptor
- High affinity
- Modeled on CP0597 by replacing �-turn conformation of the peptide by a rigid 1,4 piperazine ring
CP2522
- Human B2 receptor
- B2 receptor antagonist at the nanomolar range
Substituted 1,4-dihydropyridines
- Rodent
- Hypertension, inflammation
Bradyzide
- Alkakoid isolated from the South American tropical plant Martinella iquitosensis
- Affinity for both B1 and B2 receptors at the micromolar range but not selective
Nnatural compound
Pyrroloquinoline alkaloid: Martinelline
- Complex metabolite isolated from a culture of the mould Microsphaeropsis sp.
- No future pharmacological data
- Inhibition of bradykinin binding to cloned human B2 receptor at micromolar range
L-755807
Table 1: Agonists and antagonists of B2 receptors Ligands application studies.
Bradykinin B2 receptor agonists
By stimulation of BK B2 receptors, kinins exert a wide variety of diverse biological actions [6]. In various humans and animals studies, it has been demonstrated that the stimulation of BK B2 receptors is implicated in cardioprotective mechanism [6,12]. The best-characterized peptide BK B2 receptor agonists are JMV-1116 and RMP-7 [13]. The key feature of these two potent and selective agonists is their improved resistance to enzymatic degradation by kininases, rendering them valuable pharmacological tools for studying the effects of BK B2 receptor stimulation without the accompanying rapid breakdown of BK [6]. In vitro study of JMV- 1116 exhibited a high affinity towards the human cloned B2 receptor in a radioligand-binding assay [13]. Moreover, JMV-1116 behaved as a full BK B2 receptor agonist on human umbilical vein and rat uterus preparation with the same efficacy as BK [13]. Preliminary clinical trials in oncology indications confirmed that RMP-7 permeabilised the blood-brain tumour barrier to increase the delivery of agents such as carboplatin to tumors [14]. Moreover, RMP-7 enhances drug delivery to solid peripheral tumors [14,15]. The first potent, selective non-peptide BK B2 receptor agonists FR-190997 and FR- 191413 were discovered in 1997 [6,12,16]. FR-190997 is described as 4-(2-pyridyl-methoxy)-quinolone and FR-191413 is described as 3-(2-pyridylmethoxy)-benzimidazole [6,12]. The structures of both these two BK B2 receptor agonists are closely related to those of non-peptide BK B2 antagonists such as quinolone FR-167344 and benzimidazole FR-173657 [12]. The key difference between BK B2 receptor antagonist and agonist is the substitution of the quinolone moiety in position 4 with an additional 2-pyridylmethoxy group in the case of FR-190997 and the substitution of the benzimidazole moiety in position 3 with an additional 2-pyridylmethoxy group in the case of FR-191413 [6,12].
The 4-(2-pyridylmethoxy)-quinolone is a very potent nonpeptide BK B2 receptor agonist [6,12]. FR-190997 was reported to stimulate IP3 hydrolysis in Chinese Hamster Ovary (CHO) cells transfected with the human BK B2 receptor and to increase PGE2 production in human fibroblast [6,12]. However, detailed in vitro evaluations on human, rabbits and pig vascular BK B2 receptors have revealed that this agonist shows different intrinsic activity depending on the tissue preparations [16]. FR-190997 acts as a partial agonist in the human umbilical vein and especially in the rabbit jugular vein, but as a pure antagonist in the pig coronary artery [16]. From this study it was suggested that the addition of a 2-pyridylmethoxy group to the basic structure of the antagonist FR-173657 might not be completely sufficient to change the pharmacological spectrum of a pure antagonist to that of a full agonist [16]. FR-190997 induced hypotensive response in anaesthetized spontaneously hypertensive rats after continuous infusion into the abdominal aorta [17]. Moreover, FR-190997 induced natriuresis and diuresis without any influence on urinary potassium excretion in anaesthetized rabbits [18].
FR-191413 is a second promising series of non-peptide BK B2 receptor agonists. The pharmacological properties of FR-191413 have been less intensively evaluated than those of FR-190997 [6]. FR-191413 was reported to have similar effects to those obtained for FR-190997 on membranes CHO cells tranfected with the human BK B2 receptor. This indicates that FR-191413 is almost equipotent to FR-190997 in vitro [12]. Potent non-peptide BK B2 agonists have been discovered recently, but the oral efficacy of these agonists is very limited due to their inadequate pharmacokinetic properties [6]. This excludes their development as potential therapeutic agents for the treatment of all cardiovascular indications [6].
The Kinin System in Cardiovascular Disorders
Hypertension
Hypertension is a major risk factor for the development of cardiovascular diseases, for example, coronary heart disease, congestive heart failure, and peripheral vascular and renal diseases [1,2,5]. Kinins have potent diuretic and natriuretic effects that regulate sodium excretion from the kidney [6]. Moreover, kinins have a vasodilator action on peripheral blood vessels [1,2,5,6]. The deficiency of KKS may participate in the genesis of hypertension [2]. The involvement of kinins in blood pressure regulation has been confirmed in transgenic mouse models overexpressing human BK B2 receptor [19]. In a transgenic mouse model, over-expression of BK B2 receptor caused the development of sustained lifetime hypotension. Administration of aprotinin, a tissue kallikrein inhibitor, or icatibant, a specific B2 receptor antagonist, to the transgenic mouse restored their blood pressure to normal levels [19]. The suppression of the hypotensive response of ACE inhibitors by aprotinin in spontaneously hypertensive rats has been documented [20]. Research on the systemic changes in the KKS has provided further insight regarding various hypertensive conditions [1,5]. In essential and malignant hypertension, it is known that kininogen levels and kininpotentiating factor are reduced [1,2,5]. It may be possible that the deficiency in plasma HMWK is due to decreases in liver synthesis in individuals who develop hypertension after mild exercise [1,2]. It seems possible that deficient kallikrein-kininogens-kinin formation might be a significant factor in pathophysiology of hypertension. Renal KKS may cause excretion of excessive sodium in the urine [1,2,5,6]. Studies on the role of the renal KKS using congenitally kininogen-deficient Brown-Norway Katholiek rat and also BK B2 receptor knockout mice revealed that this system induces natriuresis and diuresis in the presence of high sodium in the body that might be due to excess sodium intake or excessive aldosterone release [21]. Thus, it can be hypothesized that the renal KKS works as a safety valve for sodium accumulation [21]. This may help in development of a compound having renal kallikrein-like activity to excrete the excess amount of sodium [1,5]. Recently, it has been proposed that tissue kallikrein gene delivery into various hypertensive models exhibit protection, for example, a reduction in high blood pressure, attenuation of cardiac hypertrophy, inhibition of renal damage and fibrosis and enhances capillary growth in spontaneously hypertensive rats [22]. These findings may indicate the prospect of this kallikrein gene therapy for cardiovascular and renal pathology [1,5]. Kininase II (ACE) inhibitors like captopril and enalapril are currently used in treatment of hypertension [1,2,5]. Kininase II inhibitors lower blood pressure by blocking the conversion of Ang I to Ang II, and increase levels of BK [2]. Abnormality in the urinary kallikrein excretion has been corrected after nifedipine, a calcium channel blocker, treatment of patients with essential hypertension [1,2]. It is a generally accepted view that the BK-induced blood pressure lowering effect is mediated by the B2 receptor, but the B1 receptor may also be involved under special situation [1]. It has been demonstrated that B2 receptor antagonist FR-173657 significantly abolished the hypotensive action of captopril [2]. Hence, it would be reasonable to suggest that the hypotensive response of ACE inhibitors is mediated mainly via B2 receptors activation [1,2]. The accumulation of BK after administration of ACE inhibitors with subsequent release of endothelium derived relaxing factor NO, PGE2 and PGI2 could be additional mediators released in the process of anti-hypertensive effects of captopril like drugs [1,2,5]. However, the use of BK antagonists can abolish the effectiveness of anti-hypertensive drugs, therefore these drugs must be contraindicated in patients with hypertension [1,5].
Cardiac Failure and Ischaemia
Cardiac failure and ischaemia are characterized by narrowed or blocked arteries, which impair the supply of blood and starve tissues of necessary nutrients and oxygen [4]. Cardiac failure and ischaemia are the leading cause of death in the developing and developed countries [1,4,5]. Kinins increase coronary perfusion and reduce preload, afterload and oxidative stress via stimulation of the release of NO and PGs mainly from the coronary cardiac endothelium, which protects the heart from acute ischaemic damage [1,5,6]. But the possible effects of kinins in the heart have been neglected for a long time [1,4,5]. ACE not only acts as a kinin-degrading enzyme but also contributes to Ang II generation [4]. Preventing the formation of Ang II could limit ventricular dilation, delay the progression of clinical symptoms, and improve mortality rate [1,5]. The ability of ACE inhibitors to prevent kinin-degrading enzyme represents a relevant mechanism contributing to cardioprotection [1,4,5]. This concept fuelled many studies and evidence demonstrating the presence of a local KKS in the heart [1,4]. Kinins released from the vascular endothelium activate BK B1 and B2 receptors on endothelial and vascular smooth muscle cells [4,5]. Binding to B1 and B2 receptors conjointly promotes the formation of PGs and NO, which increases intracellular cAMP and cGMP concentration [4]. Thus, exerting vasodilator, anti-ischaemic and anti-proliferative effects, and preserving myocardial stores of energy-rich phosphate and glycogen [4,5]. Circumstantial evidence also suggests that a dysfunctional KKS may contribute to the pathogenesis of heart failure [1,4,5]. In fact, reduced kinin outflow from the heart and blunted NO formation have been reported in microvessels of failing human heart [1,5]. Icatibant (HOE 140) is the most potent, stable and long lasting specific B2 kinin receptor antagonist [3]. It represents a second generation of kinin antagonists, and has been a useful tool to evaluate the role of kinins in cardiac ischaemia [3]. In isolated rat hearts, perfusion with HOE 140 caused reversal of the beneficial BK cardioprotective effects and facilitated the development of cardiac failure [3]. Kinins act as a cardioprotective agent in perfusion and participate in the process of ischaemic preconditioning. Preconditioning is a protective adaptive mechanism produced by short periods of ischaemic stress; this protection includes reduction in ischaemic cellular damage and in life-threatening ventricular arrhythmias [3]. BK infused into the coronary artery of anesthetized dogs reduces significantly, the severity of ischaemiainduced arrhythmia [3,5]. Rats, dogs and human studies revealed that kinins are released under the conditions of ischaemia and myocardial infarction [3]. This will help in determining the role of kinin in protecting the heart under myocardial infarction. After myocardial infarction, increased plasma levels of kallikrein, kininogen and BK were found, and the increase in plasma kallikrein levels was positively correlated with early survival rate of post-myocardial infarction patients [23,24]. It has been shown that kinins are released directly from the myocardium during myocardial infarction and contribute to the impact of ischaemic damage [23,24]. Moreover, the influence of the BK B2 receptor on the infarct size in rats with permanently ligated coronary arteries has already been confirmed using a nonpeptide BK B2 receptor antagonist and agonist, respectively. In this model, the infarct size was reduced by a continuous infusion of the BK B2 receptor agonist FR-190997, whereas it was increased upon oral administration of the BK B2 receptor antagonist FR-173657 [25]. The effect of locally administered BK on the limitation of infarct size has been investigated. In anesthetized dogs, the left descending coronary artery was ligated for 6 hours. One group received saline into the main stem of the left coronary artery. The second group received BK in a subhypotensive dose of 1ng. Kg –1. min –1. The intracoronary route and the very low dose of BK were chosen to obtain a local cardiac effect with no or minimal effects on systemic hemodynamics. Within 6 hours of coronary occlusion, infusion of BK had no significant effect on systemic blood pressure [3]. This study showed that BK limited infarct size and provided evidence for the involvement of kinins in ischaemic events [3]. In conclusion, kinins may act as mediators of endogenous cardioprotective agents. Kinins are generated and released during ischaemia, with subsequent formation of PGs and NO probably derived mainly from the coronary vascular endothelium. Their cardioprotective profiles resemble those of ACE inhibitors. However, more investigations are needed on the molecular biology and gene mapping of KKS in the heart during health and cardiovascular disorders, which may provide the answers to many questions about the role of KKS in cardiovascular pathophysiology [1,5].
Myocardial hypertrophy
Left Ventricular Hypertrophy (LVH) has been shown to be an independent predictor for increased morbidity and mortality from cardiac diseases [1,5,6]. The antihypertrophic efficacy of BK is due to B2 receptor mediated release of the endothelial cardioprotective NO and PGs [5,6]. However, B2 receptor antagonists and NO synthetase inhibitors can counter this effect [1,5]. In this regard, in hypertensive and diabetic rats, the lack of cardiac kinin forming components in the development of LVH has been documented [26]. Therefore, reduced cardiac tissue kallikrein and cardiac kininogen may be responsible for reduced BK generation in the heart that will lead to LVH [1,5]. In more recent study, in adult rat isolated cardiomyocytes and in Langendorff-perfused rat hearts, it was shown that BK exerts a direct inhibitory action against the acute hypertrophic response to Ang II in rat isolated hearts, and elevation of cardiomyocyte cGMP may be an important anti-hypertrophic mechanism caused by BK in the heart [27]. In another recent study in hypertensive rats, blood pressure reduction and regression of LVH with captopril treatment might be due to enhanced renal tissue kallikrein activity [28]. This may further support the view that tissue kallikrein may act as a cardioprotective agent [1,5].
The most conclusive evidence suggests that BK 2 receptor activation limits the infarct size and lowers the threshold for myocardial preconditioning of angiotensin converting enzyme inhibitor treatment in the isolated rat heart [29]. This observation supports the view that BK 2 receptor agonists may have future prospect of treating cardiovascular disorders. Also, BK is able to induce mitochondrial reactive oxygen, cGMP, protein kinase G and mitochondrial ATPsensitive K+ channel opening to cause cardioprotection in the rabbit cardiomyocytes [30].
It is of interest to learn that BK inhibits development of myocardial infarction through BK2 receptor signalling by increment of regional blood flow around the ischaemic lesion in the rats [31]. Furthermore, BK at reperfusion reduces infarction in rabbit hearts through PI3K, ERK and NO [32]. It has been indicated that the mechanical load caused by pressure-overload produces a down-regulation of BK 2 receptor expression during the initial stage of LVH [33].
Thus, these recent findings may highlight the significance of the BK is the protection of heart in the various disorders. Most recently, a discovery of the first non-peptide full agonist for human BK 2 receptor development in Japan [34] may provide us with the greater prospects of the application of this agonist in experimental and clinical values in cardiology.
The unwanted effects of kinin agonists
Kinin agonist displayed very pronounced and long-lasting pro-inflammatory properties in vivo [6,12]. It increased vascular permeability upon intradermal injection into dorsal skin of rats in a dose-dependant, long-lasting manner; markedly induced paw swelling upon subcutaneous injection into the hind paw of mice; enhanced frequency of writhing reactions in mice upon intraperitoneal injection in mice; and increased angiogenesis as well as granulation upon topical application in mice sponge implants [17]. BK B2 receptor-mediated activation of cyclooxygenase COX- 1 or COX-2 and synergism with PGI2 are suggested as underlying mechanisms of the described events [6]. Interestingly, FR-190997 proved to be a significant less active bronchoconstrictor than BK [17]. These observations indicate that pain induction or edema formation should be considered as one of the peripheral side effects of stable kinin receptor agonists when they have developed therapeutic usefulness [6,12]. Therefore, further studies are required to demonstrate that there is a sufficient therapeutic window between the pathophysiological and beneficial kinin receptor-mediate effects, which would allow the treatment of inflammatory disorders and chronic pain with BK B2 receptor antagonists without diminution of the beneficial systemic and cardiovascular effects of endogenous kinins and, vice versa, the treatment of cardiovascular and metabolic diseases with BK B2receptor agonists without concomitant proinflammatory side effects, especially upon chronic administration [6,12].
Conclusion
Recent studies indicate that the KKS has a role in the various pathophysiological process of the cardiovascular system, such as in hypertension, cardiac failure and ischaemia and myocardial hypertrophy. It seems that the activity of the KKS in the pathological conditions of hypertension, cardiac failure and the development of myocardial hypertrophy is deficient. These pathological states may be due to genetic abnormality of the KKS or to down regulation of BK receptors. These diseases may be treated by the application of BK B2 receptor agonists. However, these hypothesis can only be confirmed when the defined roles of KKS have been fully established after more preclinical therapeutic investigations on human.
Recent studies indicate that BK B2 receptor agonists may offer therapeutically valuable novel opportunities for the treatment and even prevention of cardiovascular diseases. However, BK B2 receptor agonists have to demonstrate efficacy in cardiovascular diseases without potential concomitant proinflammatory side effects, especially upon chronic administration. In conclusion, BK B2 receptor agonist represents a promising but challenging pharmacological target for the treatment of a wide spectrum of cardiovascular diseases.
References
- Sharma JN, Sharma J. Cardiovascular properties of the kallikrein-kinin system. Curr Med Res Opin. 2002; 18: 10-17.
- Sharma JN, Uma K, Noor AR, Rahman AR. Blood pressure regulation by the kallikrein-kinin system. Gen Pharmacol. 1996; 27: 55-63.
- Linz W, Wiemer G, Schölkens BA. Role of kinins in the pathophysiology of myocardial ischemia. In vitro and in vivo studies. Diabetes. 1996; 45 Suppl 1: S51-58.
- Emanueli C, Madeddu P. Targeting kinin receptors for the treatment of tissue ischaemia. Trends Pharmacol Sci. 2001; 22: 478-484.
- Sharma JN. Does the kinin system mediate in cardiovascular abnormalities? An overview. J Clin Pharmacol. 2003; 43: 1187-1195.
- Heitsch H. The therapeutic potential of bradykinin B2 receptor agonists in the treatment of cardiovascular disease. Expert Opin Investig Drugs. 2003; 12: 759-770.
- Campbell DJ. The kallikrein-kinin system in humans. Clin Exp Pharmacol Physiol. 2001; 28: 1060-1065.
- Yayama K, Nagaoka M, Takano M, Okamoto H. Expression of kininogen, kallikrein and kinin receptor genes by rat cardiomyocytes. Biochim Biophys Acta. 2000; 1495: 69-77.
- Campbell DJ. Towards understanding the kallikrein-kinin system: insights from measurement of kinin peptides. Braz J Med Biol Res. 2000; 33: 665-677.
- Regoli D, Rizzi A, Perron SI, Gobeil F Jr. Classification of kinin receptors. Biol Chem. 2001; 382: 31-35.
- Agata J, Miao R, Yayama K, Chao L, Chao J. Bradykinin B(1) receptor mediates inhibition of neointima formation in rat artery afeter balloon angioplasty. Hypertension. 2000; 36: 364-370.
- Heitsch H. Non-peptide antagonists and agonists of the bradykinin B(2) receptor. Curr Med Chem. 2002; 9: 913-928.
- Amblard M, Daffix I, Bedos P, Bergé G, Pruneau D, Paquet JL, et al. Design and synthesis of potent bradykinin agonists containing a benzothiazepine moiety. J Med Chem. 1999; 42: 4185-4192.
- Shimuta SI, Barbosa AM, Borges AC, Paiva TB. Pharmacological characterization of RMP-7, a novel bradykinin agonist in smooth muscle. Immunopharmacology. 1999; 45: 63-67.
- Emerich D. Use of bradykinin agonist, cereport, as a pharmacological means of increasing drug delivery to the brain. Curr. Med. Chem. Immunol. Endocrine Metabol. Agents. 2002; 2: 109-123.
- Rizzi A, Rizzi C, Amadesi S, Calo G, Varani K, Inamura N, et al. Pharmacological characterization of the first non-pepetide bradykinin B2 receptor agonist FR-190997: an in vitro study on human, rabbit and pig vascualar B2 receptors. Naunyn Schmiedebergs Arch Pharmacol. 1999; 360: 361-367.
- Ueno A, Naraba H, Kojima F, Morita E, Ohishi S. FR-190997, anovel bradykinin B2 agonist, expresses longer action than bradykinin in paw edema formation and hypotensive response. Immunopharmacology. 1999; 45: 89-93.
- Majima M, Hayashi I, Inamura N, Fujita T, Ogino M. A nonpeptide mimic of bradykinin blunts the development of hypertension in young spontaneously hypertensive rats. Hypertension. 2000; 35: 437-442.
- Wang D, Yoshida H, Song Q, Chao L, Chao J. Enhanced renal function in bradykinin B(2) receptor transgenic mice. Am J Physiol Renal Physiol. 2000; 278: F484-491.
- Sharma JN, Amrah SS, Noor AR. Suppression of hypotensive responses of captopril and enalapril by the kallikrein inhibitor aprotinin in spontaneously hypertensive rats. Pharmacology. 1995; 50: 363-369.
- Katori M, Majima M. The renal kallikrein-kinin system: its role as a safety valve for excess sodium intake, and its attenuation as a possible etiologic factor in salt-sensitive hypertension. Crit Rev Clin Lab Sci. 2003; 40: 43-115.
- Bledsoe G, Chao L, Chao J. Kallikrein gene delivery attenuate cardiac remodeling and promotes neovascularization in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2003; 285: H1479-1488.
- Tschöpe C, Heringer-Walther S, Walther T. Regulation of the kinin receptors after induction of myocardial infarction: a mini-review. Braz J Med Biol Res. 2000; 33: 701-708.
- Tschöpe C, Heringer-Walther S, Koch M, Spillmann F, Wendorf M, Hauke D, et al. Myocardial bradykinin B2-receptor expression at different time points after induction of myocardial infarction. J Hypertens. 2000; 18: 223-228.
- Ito H, Hayashi I, Izumi T, Majima M. Bradykinin inhibits development of myocardial infarction through B2 receptor signalling by increment of regional blood flow around the ischaemic lesions in rats. Br J Pharmacol. 2003; 138: 225-233.
- Sharma JN, Uma K, Yusof AP. Left ventricular hypertrophy and its relation to the cardiac kinin-forming system in hypertensive and diabetic rats. Int J Cardiol. 1998; 63: 229-235.
- Rosenkranz AC, Hood SG, Woods RL, Dusting GJ, Ritchie RH. Acute antihypertrophic actions of bradykinin in the rat heart: importance of cyclic GMP. Hypertension. 2002; 40: 498-503.
- Sharma JN, Kesavarao U. Effect of captopril on urinary kallikrein, blood pressure and myocardial hypertrophy in diabetic spontaneously hypertensive rats. Pharmacology. 2002; 64: 196-200.
- Ebrahim Z, Baxter GE, Yellon DM. Omapatrilat limits infarct size and owers the threshold for induction of myocardial preconditioning through a bradykinin receptor –mediated mechanism. Cardiovasc. Drugs. Ther. 2004; 18: 127-134.
- Oldenburg O, Qin Q, Krieg T, Yang XM, Philipp S, Critz SD, et al. Bradykinin induces mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP channel opening and leads to cardioprotection. Am J Physiol Heart Circ Physiol. 2004; 286: H468-476.
- Ito H, Hayashi I, Izumi T, Majima M. Bradykinin inhibits development of myocardial infarction through B2 receptor signalling by increment of regional blood flow around the ischaemic lesions in rats. Br J Pharmacol. 2003; 138: 225-233.
- Yang XM, Krieg T, Cui L, Downey JM, Cohen MV. NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3K, ERK, and NO. J Mol Cell Cardiol. 2004; 36: 411-421.
- Yayama K, Matsuoka S, Nagaoka M, Shimazu E, Takano M, Okamoto H. Down-regulation of bradykinin B2-receptor mRNA in the heart in pressure-overload cardiac hypertrophy in the rat. Biochem Pharmacol. 2003; 65: 1017-1025.
- Sawada Y, Kayakiri H, Abe Y, Mizutani T, Inamura N, Asano M, et al. Discovery of the first non-peptide full agonists for human bradykinin B(2) receptor incorporating 4-(2-picolyloxy)quinolin and 1-(2-picolyl)benzimidazole frameworks. J. Med. Chem. 2004; 47: 2853-2863.