
Review Article
A Proteomics. 2014;1(1): 7.
Applications of Formalin Fixed Paraffin-Embedded Tissue Proteomics in the Study of Cancer
Juliana de S da G Fischer1, Paulo C Carvalho1, Nathalie HS Canedo2, Katia Maria da S Gonçalves2, Priscila Ferreira Aquino1,3, Barbara Almeida2, Vera LN Pannain2, and Maria da Gloria C Carvalho2*
1Laboratory for proteomics and protein engineering, Carlos Chagas Institute, Fiocruz - Paraná, Brazil
2Department of Pathology, University Hospital Clementino Fraga Filho, Federal University of Rio de Janeiro, Brazil
3Chemistry Institute, Federal University of Rio de Janeiro, Brazil
*Corresponding author: Maria da Gloria da Costa Carvalho, Department of Pathology, University Hospital Clementino Fraga Filho, Federal University of Rio de Janeiro, Rua Prof. Rodolpho Rocco 255, ZIP 21941-913 Rio de Janeiro-RJ, Brazil. CEP: 22421-020
Received: June 20, 2014; Accepted: July 31, 2014; Published: Aug 05, 2014
Abstract
Among the many challenges in identifying novel tumoral diagnostic, prognostic, and personal therapy biomarkers we highlight the challenge of dealing with a single tumor's molecular complexity and heterogeneity. Formalin-fixed paraffin-embedded (FFPE) tissue specimens are commonly used by pathologists for diagnosing cancer; this material comprises a valuable resource for molecular studies addressing tumoral molecular profiles. Here we demonstrate the effectiveness of shotgun proteomics in assessing the cellular heterogeneity using FFPE tissues slides under three different scenarios: 1) Comparing astrocytoma grade I versus glioblastoma, 2) Assessing different morphological areas from the same histological slide of a glioblastoma and comparing the identifications with those from a fresh tissue, 3) Comparing two different histopathological profiles in the same patient's liver: cirrhosis and cancer. Briefly, contents of FFPE slides were scraped, washed three times with xylene and then rehydrated in a grade ethanol series [100%, 90%(v/v), 70 % (v/v)]. A total of 100 μL of 0.2% (w/v) RapiGest™ in 50mM ammonium bicarbonate was then added to each sample. Tryptic digests were separated by reversed-phase capillary liquid chromatography coupled to nano-electrospray high-resolution mass spectrometry for identification. The samples were analyzed in technical replicates by LC-MS/MS on an Orbitrap XL. Our datasets enabled elucidating key tumoral regions allowing a better comprehension of such specific tumor areas; the identifications and raw data are available at https://max.ioc.fiocruz.br/ julif/ffpecases.
Keywords: Formalin fixed paraffin-embedded; Glioblastoma; Hepatic cancer; Cirrhosis; Shotgun proteomics
Introduction
Tumors consist of cells in different stages of transformation, resulting in both molecular and cellular heterogeneity. While cellular morphology is characterized by pathologist, it is now known that what might apparently seems as morphologically healthy tissue might already be molecularly compromised [1,2]. The application of mass spectrometry based proteomics in the analysis of formalin fixed paraffin-embedded (FFPE) tissue poses as a powerful technique to enable an in depth study of specific tumoral areas [3]. Needless to say, there is a plethora of FFPE tissue specimens available, which composes a treasure-trove for retrospective studies paving the way for mass spectrometric assessment of archived material of rare diseases [4].
To exemplify the usefulness of FFPE in molecularly assessing tumoral heterogeneity we demonstrate applications of FFPE proteomics by studying three very distinct scenarios: in the first, we compare the relationship between astrocytoma grade I and glioblastoma; in the second, different morphological regions from the same glioblastoma histological slide and a fresh GBM tissue sample are contrasted; the last compares two different histopathological areas, cirrhosis and hepatic cancer, from the same patient.
Methods
The sample preparation and data analysis for FFPE cancer proteomics performed for generating the datasets are described henceforth. The ethics committee review board of the University Hospital Clementino Fraga Filho reviewed and approved this work under number 060-11.
FFPE protein extraction
The contents of microscope slides were scraped using a syringe needle and the material transferred to an Eppendorf type tube. Then the contents were washed three times with 250 μL of xylene at 60°C. Afterwards, the contents were rehydrated with 250 μL of a grade ethanol series [100%, 90%(v/v), 70 % (v/v)], followed by centrifugation during 30 minutes at 15000 x g and air dried at room temperature until complete ethanol evaporation.
A total of 100 μL of 0.2% (w/v) RapiGest TM in 50mM ammonium bicarbonate was then added to each sample. The protein mass within the samples was estimated using the BCA protein assay Kit (Sigma- Aldrich) according to the manufacturer's instructions. The contents from each sample were reduced with 5mM of dithiothreitol (DTT) and incubated for 90 min at 95°C, followed to 2 h of incubation at 70°C. The sample incubation is necessary to hydrolyze the cross links generated during formalin fixation of the tissue. The samples were cooled to room temperature and incubated in the dark with 15mM of iodoacetamide (IAA) for 30 minutes.
Fresh GBM protein extraction
A fresh GBM sample was obtained from a 71 year old man with no family history of brain tumor. The man began to feel strong headaches and would not respond to analgesics. A brain magnetic resonance imaging scan (MRI) revealed a lesion in the left temporal lobe. After signing informed consent, the patient underwent left temporal biopsy and was diagnosed with glioblastoma multiform according to three independent pathologists.
The glioblastoma tissue lysate was obtained by sonication ((Dr. Hielscher) with amplitude of 70% and 3 cycles of one minute of the biopsy immerse in a solution containing 0.2% of RapiGest (w/v) in 50mM ammonium bicarbonate. Then, the solution was centrifuged at 15000 x g during 30 minutes at 5oC. The supernatant was transferred to a new tube and the protein content was estimated using the BCA protein assay Kit (Sigma-Aldrich) according to the manufacturer's instructions. One hundred micrograms of protein were reduced with 20mM dithiothreitol (DTT) at 60°C for 30 minutes. After that, samples were cooled to room temperature and incubated, in the dark, with 66mM of iodacetamide (IAA) for 30 minutes.
Protein digestion and mass spectrometry analysis
Afterwards, all samples were digested overnight with trypsin (Promega) at the proportion of 1/50 (E/S). Following digestion, all reactions were acidified with 10% (v/v) formic acid (1% (v/v) final) to stop the proteolysis. The samples were centrifuged for 20 minutes at 15,000 x g to remove insoluble material and desalted with stage-tips prior the RP - LC Mass spectrometry. The desalted peptide mixture was subjected to LC-MS/MS analysis with a Thermo Scientific Easy-nlC 1000 ultra high performance liquid chromatography (UPLC) system coupled with a LTQ-Orbitrap [5] XL ETD (Thermo, San Jone, CA) mass spectrometer described as follows. The peptide mixtures were loaded onto a column (75 mm i.d., 15 cm long) packed in house with a 3.2 μm ReproSil-Pur C18-AQ resin (Dr. Maisch) with a flow of 500 ml/min and subsequently eluted with a flow of 250ml/min from 5% to 40% ACN in 0.5% formic acid, in a 120 min gradient. The mass spectrometer was set in data dependent mode to automatically switch between MS and MS/MS (MS2) acquisition. Survey full scan MS spectra (from m/z 300 - 2000) were acquired in the Orbitrap analyzer with resolution R = 60,000 at m/z 400 (after accumulation to a target value of 1,000,000 in the linear trap). The nine most intense ions were sequentially isolated and fragmented in the linear ion trap using collisional induced dissociation at a target value of 10,000. Previous target ions selected for MS/MS were dynamically excluded for 90 seconds. Total cycle time was approximately three seconds. The general mass spectrometric conditions were: spray voltage, 2.4 kV; no sheath and auxiliary gas flow; ion transfer tube temperature, 100°C; collision gas pressure, 1.3 mTorr; normalized energy collision energy using wide-band activation mode; 35% for MS2. Ion selection thresholds were of 250 counts for MS2. An activation q = 0.25 and activation time of 30 ms was applied in MS2 acquisitions.
The mass spectra were extracted to the MS2 format using Pattern Lab's Raw Reader. Sequences from Homo sapiens were downloaded from the UniProt consortium on November 13th, 2013. A target decoy database was generated using Pattern Lab to include a reversed version of each sequence found in the database plus those from 127 common mass spectrometry contaminants. The ProLuCID search engine (v1.3) [6] as limited to fully and semi-tryptic peptide candidates; we imposed carbamidomethylation and oxidation of methionine as a fixed and variable modifications, respectively. The search engine accepted peptide candidates within a 70-ppm tolerance from the measured precursor m/z and used the XCorr as the primary search engine score. The Search Engine Processor (SEPro) (v2.2.0.2) was used for achieving a less than 1% FDR at the protein level as previously described [7]. PatternLab's Approximately Area Proportional Venn Diagram module was used to pinpoint proteins uniquely identified in each case [8]. All proteins discussed along the text and claimed as uniquely identified in a biological condition were found in at least two technical replicates (except for case 1 that contains single replicates due to limited material). As previously discussed, the more a protein appears in replicates of a biological condition and remains absent in the other, the more likely it is to be differentially expressed or unique to that condition [9].
Results and Discussion
Scenario 1: Astrocytoma grade I versus glioblastoma
The first scenario compares astrocytoma grade I and glioblastoma from microscope slides of formalin fixed paraffin-embedded tissues. To evaluate the differentially expressed proteins in astrocytoma grade I (pilocytic astrocytoma) and grade IV (glioblastoma), the contents of five 5 μm sections obtained from each case were scraped from the microscope slides and the proteins were extracted and analyzed once as described above. The microscope slide images stained with hematoxylin and eosin are shown in Figure 1. Our results describe 715 (145 by maximum parsimony [10]) and 623 (110 by maximum parsimony) proteins identified from astrocytoma grade I and glioblastoma section, respectively.
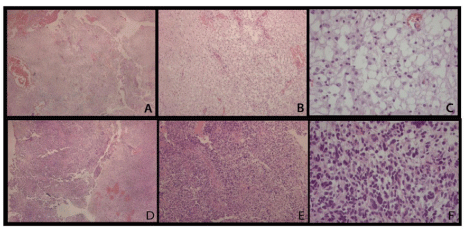
Figure 1 : Hematoxylin and eosin stained slides from astrocytoma grade I (A, B, C) and glioblastoma (D, E, F). A, D: 40x; B, E: 100x; C, F: 400x. In image C, the characteristic microcystic areas are prominent and little pleomorphism can be noticed, in contrast with image F, where nuclear atypia and hypercellularity are easily seen.
We used the Pattern Lab for proteomics for pinpointing identified proteins from astrocytoma grade I and glioblastoma. Our results showed that 52% of proteins were common to both grades, 256 proteins were exclusively identified in astrocytoma grade I and 164 in the glioblastoma. The total number of identified proteins was 879 (178 by maximum parsimony) when considering all results simultaneously. We recall that not identifying a protein by mass spectrometry means that it is either not present in the sample or is under the detection limits of the strategy at hand; as such, it can be hypothesized as "off" or as differentially expressed when comparing to a condition where it is identified [9]. We opted for this conservative strategy as in according to a previous report where analyses were performed in the same conditions and with very limited sample [11].
Among the proteins exclusively identified in the glioblastoma dataset, it is worth pinpointing 14-3-3 gamma, dermcidin, and defensin. Proteins of the 14-3-3 family are linked to many key cellular processes related to cancer such as apoptosis and cell cycle key points; for these reasons, they are considered as important therapeutic targets [12]. In particular, 14-3-3 gamma was previously described, among eleven other proteins, as glioblastoma biomarker candidates by the use of large scale proteomic approaches [13]. Dermcidin and defensin were both described as having antimicrobial effects; interestingly, both were only identified in the glioblastoma sample. Dermcidin and dermcidin-derived peptides have been describe to play a key role in several biological functions in neuronal and cancer cells [14]; although this protein has been described as up-regulated in various cancer cell types [14], as far as we know, this is the first report claiming such for glioblastoma. Defensin has been described as a potential strategy for cancer immunotherapy as promising immunogenes [15].
Among the proteins exclusively identified in astrocytoma grade I we highlight annexin A1 and Brevican. Annexin-1 (ANXA1) belongs to a structurally related family of calcium-binding anti-inflammatory proteins that act extracellularly as an inhibitor of icosanoid synthesis and phospholipase A2 [16]. Schittenhelm et al. proposed that the up-regulation of annexin-1 in astrocytomas is linked to a bad prognosis for tumor; moreover showed that its expression profile is highly correlated to its substrate, EGFR [17]. Brevican has been previously correlated as a marker for low-grade gliomas [18]. Figure 2 shows a carbon metabolism metabolic pathway map; as can be noticed, different sections of the pathways are up-regulated in each type of cancer.
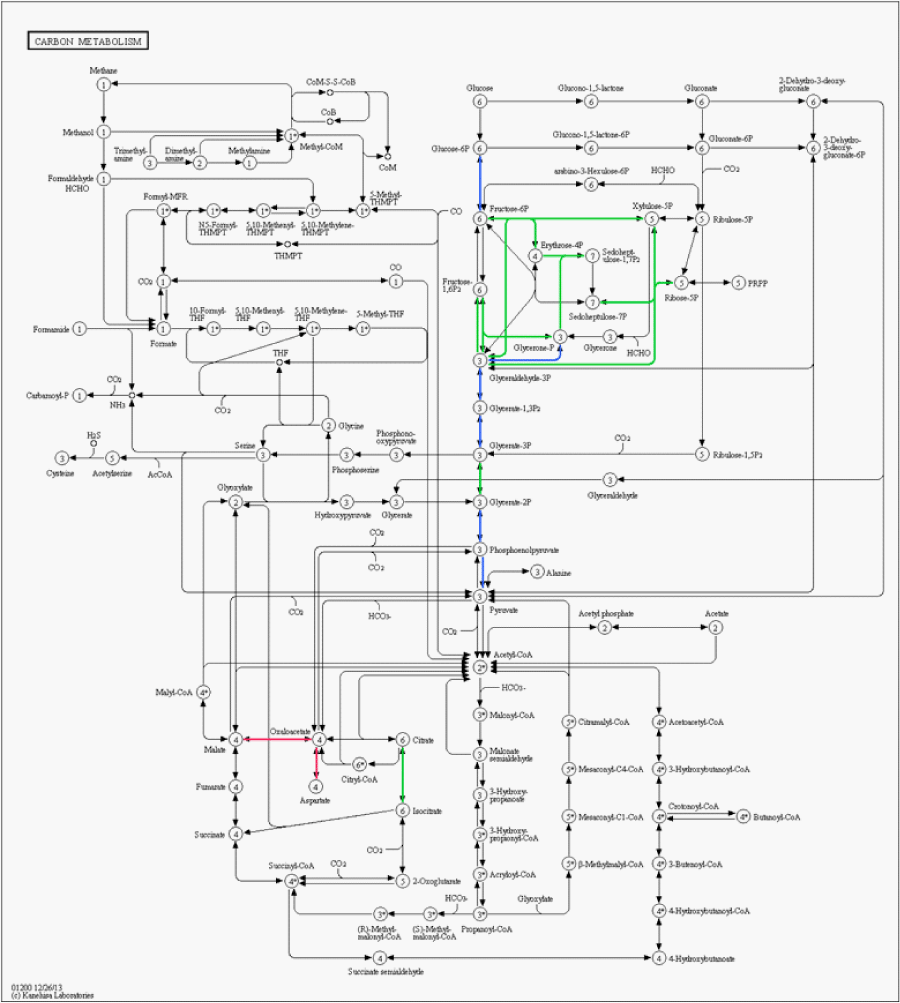
Figure 2 : Metabolic pathway map of the carbon metabolism generated using KEGG mapper. Green links are associated to up-regulated proteins in astrocytoma grade I, red links to glioblastoma, and blue links indicate no significant change in regulation.
Scenario 2: Different morphological regions from the same glioblastoma histological slide and comparison with fresh GBM tissue
The second dataset compares FFPE tissue of two distinct areas of the same glioblastoma tumor and GBM from fresh tissue; each condition had three technical replicate analyses. Figure 3 shows a picture of the histological slide used for obtaining the material for this analysis. Glioblastomas characteristically exhibit a wide range of morphological aspects, from small cell areas to giant or highly pleomorphic areas, although considered to be monoclonal in origin. Irrespectively, the clinical outcome is said to be unrelated to this variation. The goal was to verify if a proteomic assessment of FFPE could pinpoint differences in the proteomic profiles of different regions of the same tumor that presented a significantly different morphology as in accordance to a pathologist's judgment. Two regions of interest were selectively scraped from the slide and the proteins were extracted according to our methods section.
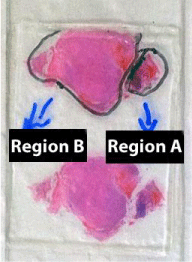
Figure 3 : A histological slide of a glioblastoma stained with hematoxylin and eosin; two regions with distinct morphology studied in the manuscript are delineated.
Our proteomic analysis revealed 1463 proteins; 439 were identified in region A (Figure 4) 1058 in region B (Figure 4) and 705 in the fresh tissue sample. We expected to identify more proteins in the fresh tissue as in according to previous works [19]; yet, our results ended up being different. We believe this was due to the fresh tissue sample originating from a different patient than that of the FFPE tissue; as previously reported, there is a tremendous variation between the proteome of different individuals [20]. As an aggravator, according to Fowler et al, "even for matched fresh tissue specimens, overlap in the identified proteins can be as low as 50% due to variations in sample preparation and under sampling of the resulting peptides" [21].
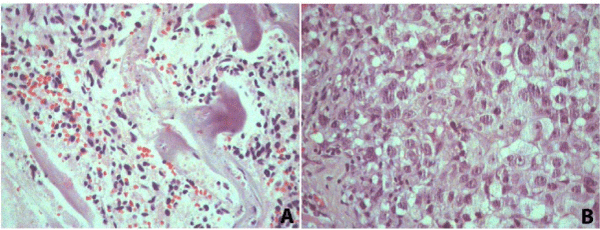
Figure 4 : Hematoxylin and eosin sections from glioblastoma. Two very distinct regions can be noticed: A- Predominantly composed by small, irregular cells. B - Containing large, pleomorphic cells. In both areas the necrosis, vascular proliferation and anaplasia were sufficient for the diagnosis of glioblastoma.
Of the 1463 proteins, 368 were identified only in the fresh tissue and another 731 were not identified in the fresh tissue. There was a significantly greater overlap in the protein identifications originating from the two different FFPE areas (92% overlap) than from FFPE versus fresh tissue (46% overlap). Details on IDs are provided at the end of this manuscript and in the supplementary table. We hypothesize that more proteins were identified in region B because more material was obtained during the protein extraction. As can be noted in Figure 3, region B is approximately 4 x larger than region A. Our results pinpoint 18 proteins exclusively identified in region A when comparing with region B. We consider this a positive result, as even though significantly fewer proteins were identified in this area, we were still able to find some "uniqueness" in it; this raises our confidence that these proteins shed important biological aspects of the disease. Such result demonstrates the great potential of this methodology to reveal the cellular heterogeneity of the tumor as reflected in the hematoxylin and eosin stains (Figure 4). Among the unique proteins identified in region A we point out cold-inducible RNA binding protein and THO complex subunit 4. The former has been described as decreased in breast cancer cells [22] and the later to have a key role in anti-cancer mechanisms [23]. That said, we emphasize that specific proteins correlate to particular histological sections and therefore could ultimately be used for molecularly defining distinct areas, as tumors are very heterogeneous.
Scenario 3: Different areas, cirrhosis and hepatic cancer, from the same patient
The third dataset compares the proteomic profiles of different pathologies in the liver of the same patient obtained in the same surgical procedure; three technical replicate analyses from each condition were performed. In this case, two different histopathological areas, cirrhosis and cancer, were identified by three independent pathologists. This makes the material at hand extremely valuable as it is hard to be obtained when belonging to the same patient. That said, this case demonstrates the usefulness of FFPE proteomics to enable retrospective studies from archival material. For the case at hand, FFPE proteomics enabled identifying proteins that could be related to the progression between these two distinct pathologies helping to elucidate the relationship between these diseases. The microscope slides were assessed by a pathologist (Figure 5); areas of necrosis were marked and excluded from our proteomic analysis.
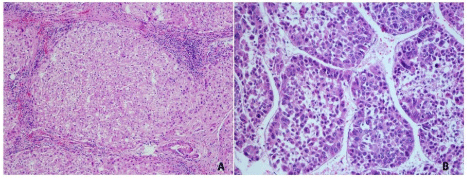
Figure 5 : Hematoxylin and eosin sections from cirrhosis (A) 100x and hepatocellular carcinoma (B) 200x.
A total of 277 proteins (69 by maximum parsimony) were identified in both cases; 115 for cirrhosis and 267 for hepatic cancer; 10 exclusively identified in cirrhosis and 162 in cancer. Among the exclusive proteins in cancer, again, the protein family of 14-3-3 poses as interesting candidate for progression biomarker to hepatic cancer. These consist a family, containing 7 isoforms of highly conserved and largely expressed eukaryotic acidic proteins (25-33kDa) that dynamically regulates the activity of target proteins in various signaling pathways and control diverse physiological and pathological processes (e.g., apoptosis and cell cycle regulation), including prion diseases [12,24,25]. 14-3-3 is integrated into the core of regulatory pathways that are key to the normal growth and development, and therefore posing this protein as progression biomarker candidate as a disturbance in these regulatory pathways can lead to cancer.
Final Considerations
A general trend in proteomics is comparing protein profiles from cancer biopsies versus those from healthy tissues ultimately entailing the discussion on how many samples are required to pinpoint candidate biomarkers. An emerging, and complementary strategy, is to dig deeper into an individual's proteome by molecularly dissecting it in association with morphological and spatial data as performed in this work. We advocate that each cancer should be considered as a disease of its own; needless to say, patients respond differently to treatment. As far as we know, the idea of associating large-scale protein identification to spatial data was pioneered by Richard Smith's group with two seminal works that sectioned a mammalian brain and produce images of identified protein distributions along the brain [26,27]. Our group's efforts are also aligned with "spatial proteomics"; we recently developed software automatically generating shotgun images and exemplified its usefulness by delineating a gastric cancer tumoral region based on Mud PIT data [28]. The literature is full of examples showing applications of FFPE for identifying candidate biomarkers through various strategies using FFPR, including Single Reaction Monitoring [29], and laser micro-dissection [30]. Clearly, there is a drive for shotgun proteomics to becoming more and more spatially centric, ultimately aiming at a single-cell's proteome [31]. In our view, studies that combine FFPE with shotgun proteomics are in a sweet-spot in the compromise between large-scale protein identification and spatial resolution; this strategy is under continuous perfection by the scientific community [32].
Here, we provide key datasets demonstrating the great potential of FFPE proteomics for identification of tumor biomarkers candidates (dataset 1), comparing distinct proteomic profiles from different regions of the same tumor versus a fresh tissue sample (dataset 2), and two distinct histopathological areas in the liver of the same patient which could possibly describe a molecular linkage between progression from cirrhosis to cancer (dataset 3). These datasets are unique as few proteomic datasets disclose shotgun identifications of tumoral specific region delineated by microscopy and with the corresponding histopathological assessment. Supplementary Table I discriminates all identifications from each biological condition of each case followed by its respective protein length (in amino acids), number of unique peptides identified, molecular weight, sequence count, spectrum count, protein coverage, number of technical replicates identified in, and the protein's description.
Figure 6 summarizes the results obtained herein using area proportional Venn diagrams. All data, including raw files and SEPro identifications are available at https://max.ioc.fiocruz.br/julif/ ffpecases/. The data is organized in three separate folders enumerated as in according to this article.
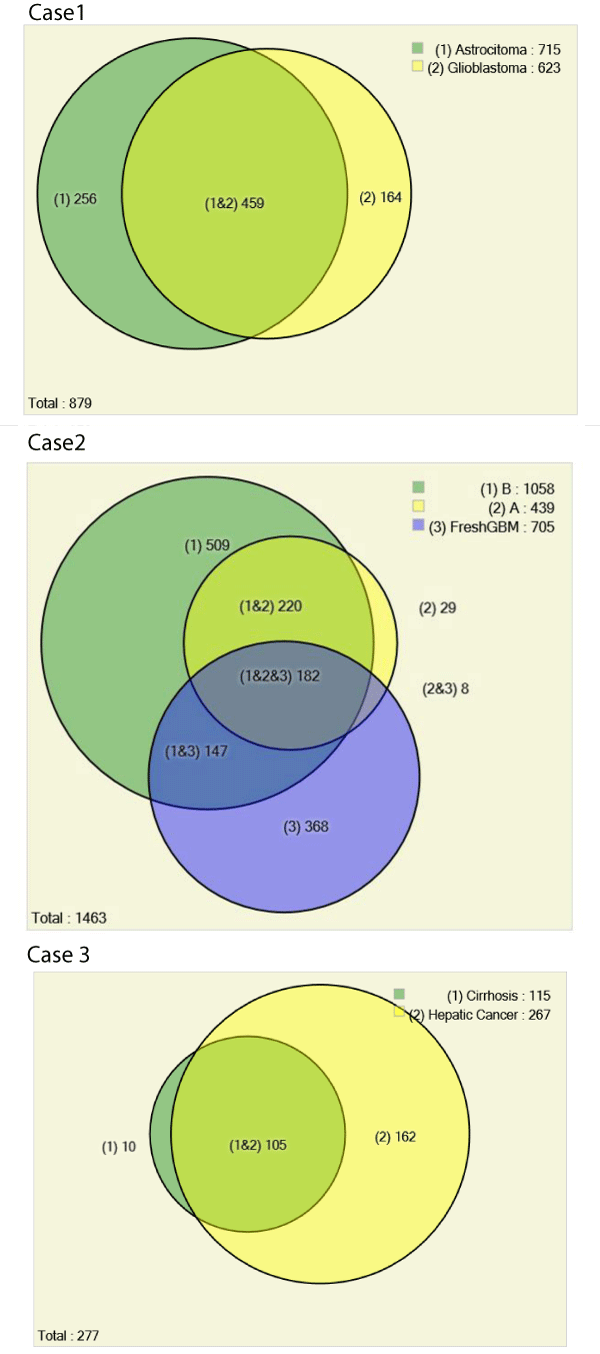
Figure 6 : Venn Diagrams comparing proteomic profiles of: astrocytoma grade I versus glioblastoma (Case 1), fresh GBM tissue and different morphological areas from the same histological slide of a glioblastoma from a different patient (Case 2), and different histopathological profiles in the same patient's liver: cirrhosis and cancer (Case 3). All proteomic identifications are decurrently from material obtained by scraping microscope slides. The results from each biological condition of case 1 were obtained from a single LC/MS/MS analysis, all other biological conditions (i.e., from cases 2 and 3) were obtained from three technical replicate analyses.
Author Contributions
JSGF, PFA, KMSG, and BA were responsible for sample preparation and helped in experimental design. PCC and MGCC wrote the manuscript and participated in discussion. PCC did the mass spectrometry data analysis. MGCC, NHSC, and VLNP are medical doctors and performed sample analysis procedures.
Acknowledgement
The authors thank CNPq, FAPERJ, and Fundação do Câncer for financial support. The authors thank the Program for Technological Development in Tools for Health-PDTIS-FIOCRUZ (PDTIS) for use of the mass spectrometry facility RPT02H PDTIS / Carlos Chagas Institute - Fiocruz Parana and the mass spectrometry core facility from Fiocruz - Rio. The authors acknowledge Dr. Michel Batista from Fiocruz Paraná and Dr. Richard Hemmi Valente from the laboratory of Toxinology, Fiocruz - Rio, for aiding in the mass spectrometry procedures. The authors declare no conflict of interests.
References
- Chaurand P, Norris JL, Cornett DS, Mobley JA, Caprioli RM. New developments in profiling and imaging of proteins from tissue sections by MALDI mass spectrometry. J Proteome Res. 2006; 5: 2889-2900.
- Kim HK, Reyzer ML, Choi IJ, Kim CG, Kim HS, Oshima A, et al. Gastric cancer-specific protein profile identified using endoscopic biopsy samples via MALDI mass spectrometry. J Proteome Res. 2010; 9: 4123-4130.
- Fowler CB, O'Leary TJ, Mason JT. Toward improving the proteomic analysis of formalin-fixed, paraffin-embedded tissue. Expert Rev Proteomics. 2013; 10: 389-400.
- Fischer Jde S, Canedo NH, Goncalves KM, Chimelli LM, Franca M, Leprevost FV, et al. Proteome analysis of formalin-fixed paraffin-embedded tissues from a primary gastric melanoma and its meningeal metastasis: a case report. Curr Top Med Chem. 2014; 14: 382-387.
- Makarov A. Electrostatic axially harmonic orbital trapping: a high-performance technique of mass analysis Anal Chem. 2000; 72: 1156-1162.
- Xu T, Venable JD, Park S, Cociorva D, Lu B, Liao L, et al. ProLuCID, a Fast and Sensitive Tandem Mass Spectra-based Protein Identification Program. Mol Cell Proteomics. 2006; 5: S174.
- Carvalho PC, Fischer JS, Xu T, Cociorva D, Balbuena TS, Valente RH, et al. Search engine processor: Filtering and organizing peptide spectrum matches. Proteomics. 2012; 12: 944-949.
- Carvalho PC, Fischer JS, Xu T, Yates JR, Barbosa VC. PatternLab: from mass spectra to label-free differential shotgun proteomics. Curr Protoc Bioinformatics. 2012; Chapter 13: Unit13.
- Carvalho PC, Fischer JSG, Perales J, Yates JR, Barbosa VC, Bareinboim E. Analyzing marginal cases in differential shotgun proteomics. Bioinforma Oxf Engl. 2011; 27: 275-276.
- Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res. 2007; 6: 3549-3557.
- Azimzadeh O, Barjaktarovic Z, Aubele M, Calzada-Wack J, Sarioglu H, Atkinson MJ, et al. Formalin-fixed paraffin-embedded (FFPE) proteome analysis using gel-free and gel-based proteomics. J Proteome Res. 2010; 9: 4710-4720.
- Hermeking H. The 14-3-3 cancer connection. Nat Rev Cancer. 2003; 3: 931-943.
- Dekker L, Kheirollahi M, Stockhammer G, Luider T, Sillevis Smitt P. Glioma Biomarker Discovery by Mass Spectrometry (P06.001). Neurology. 2012; 78: P06.001.
- Schittek B. The multiple facets of dermcidin in cell survival and host defense. J Innate Immun. 2012; 4: 349-360.
- Li D, Wang W, Shi HS, Fu YJ, Chen X, Chen XC, et al. Gene therapy with beta-defensin 2 induces antitumor immunity and enhances local antitumor effects. Hum Gene Ther. 2014; 25: 63-72.
- Lim LH, Pervaiz S. Annexin 1: the new face of an old molecule. FASEB J. 2007; 21: 968-975.
- Schittenhelm J, Trautmann K, Tabatabai G, Hermann C, Meyermann R, Beschorner R. Comparative analysis of annexin-1 in neuroepithelial tumors shows altered expression with the grade of malignancy but is not associated with survival. Mod Pathol. 2009; 22: 1600-1611.
- Viapiano MS, Bi WL, Piepmeier J, Hockfield S, Matthews RT. Novel tumor-specific isoforms of BEHAB/brevican identified in human malignant gliomas. Cancer Res. 2005; 65: 6726-6733.
- Tanca A, Pagnozzi D, Burrai GP, Polinas M, Uzzau S, Antuofermo E, et al. Comparability of differential proteomics data generated from paired archival fresh-frozen and formalin-fixed samples by GeLC-MS/MS and spectral counting. J Proteomics. 2012; 77: 561-576.
- Brunoro G, Ferreira A, Trugilho M, Oliveira T, Amêndola L, Perales J, et al. Potential Correlation between Tumor Aggressiveness and Protein Expression Patterns of Nipple Aspirate Fluid (NAF) Revealed by Gel-Based Proteomic Analysis. Curr Top Med Chem. 2014; 14: 359-368.
- Fowler CB, O'Leary TJ, Mason JT. Toward improving the proteomic analysis of formalin-fixed, paraffin-embedded tissue. Expert Rev Proteomics. 2013; 10: 389-400.
- Guo X, Wu Y, Hartley RS. Cold-inducible RNA-binding protein contributes to human antigen R and cyclin E1 deregulation in breast cancer. Mol Carcinog. 2010; 49: 130-140.
- Lin YS, Lin CH, Huang LD, Chao T, Kuo CD, Hung LC, et al. The suppression of thoc1 in cancer cell apoptosis mediated by activated macrophages is nitric oxide-dependent. Biochem Pharmacol. 2013; 86: 242-252.
- Wilker E, Yaffe MB. 14-3-3 Proteins--a focus on cancer and human disease. J Mol Cell Cardiol. 2004; 37: 633-642.
- Moreira JMA, Gromov P, Celis JE. Expression of the tumor suppressor protein 14-3-3 sigma is down-regulated in invasive transitional cell carcinomas of the urinary bladder undergoing epithelial-to-mesenchymal transition. Mol Cell Proteomics MCP. 2004; 3:410-419.
- Petyuk VA, Qian WJ, Smith RD, Smith DJ. Mapping protein abundance patterns in the brain using voxelation combined with liquid chromatography and mass spectrometry. Methods. 2010; 50: 77-84.
- Petyuk VA, Qian WJ, Chin MH, Wang H, Livesay EA, Monroe ME, et al. Spatial mapping of protein abundances in the mouse brain by voxelation integrated with high-throughput liquid chromatography-mass spectrometry. Genome Res. 2007; 17: 328-336.
- Aquino PF, Lima DB, de Saldanha da Gama Fischer J, Melani RD, Nogueira FC, Chalub SR, et al. Exploring the proteomic landscape of a gastric cancer biopsy with the shotgun imaging analyzer. J Proteome Res. 2014; 13: 314-320.
- Hembrough T, Thyparambil S, Liao WL, Darfler MM, Abdo J, Bengali KM, et al. Selected Reaction Monitoring (SRM) Analysis of Epidermal Growth Factor Receptor (EGFR) in Formalin Fixed Tumor Tissue. Clin Proteomics. 2012; 9: 5.
- Takadate T, Onogawa T, Fujii K, Motoi F, Mikami S, Fukuda T, et al. Nm23/nucleoside diphosphate kinase-A as a potent prognostic marker in invasive pancreatic ductal carcinoma identified by proteomic analysis of laser micro-dissected formalin-fixed paraffin-embedded tissue. Clin Proteomics. 2012; 9: 8.
- Albert FW, Treusch S, Shockley AH, Bloom JS, Kruglyak L. Genetics of single-cell protein abundance variation in large yeast populations. Nature. 2014; 506: 494-497.
- Kawashima Y, Kodera Y, Singh A, Matsumoto M, Matsumoto H. Efficient extraction of proteins from formalin-fixed paraffin-embedded tissues requires higher concentration of tris(hydroxymethyl)aminomethane. Clin Proteomics. 2014; 11: 4.