
Perspective
A Proteomics. 2014;1(1): 3.
New Challenges for Proteomics Technologies: A Mini Perspective Review
Yufeng Shen1*, Ljiljana Paša-Tolic2, Errol W Robinson2, Joshua N Adkins1 and Richard D Smith1
1Biological Sciences Division, Pacific Northwest National Laboratory, Richland, WA 99354, USA
2Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99354, USA
*Corresponding author: Yufeng Shen, Biological Sciences Division, and Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99354, USA
Received: September 06, 2014; Accepted: October 04, 2014; Published: October 10, 2014
Abstract
Proteomics technologies have experienced rapid advances over the last decade to identify or quantify thousands of proteins per sample, typically in a few hours, enabling proteomics applications in environmental, biological, medical, and clinical research. A number of publications have reviewed advances in proteomic technologies and applications. This short review focuses first on a discussion of sensitivity in bottom-up (i.e. digested protein) proteomics and approaches for characterization of small cell populations, and secondly on protein separations for top-down (i.e. intact protein) proteomics including discussions of key technical challenges where recent advances are elucidating specific functions of proteins in biological processes.
Keywords: Chromatography; Mass spectrometry; Ultrahigh sensitivity; Proteins and peptides; Proteomics
A proteome can be characterized by measurement of small enzymatic peptides (typically having MWs <5 kDa) from digestion of proteome proteins (i.e., "bottom-up" approach) or direct measurement of proteome proteins (e.g., MWs >5 kDa) (i.e., "top-down" approach). Bottom-up proteomics generally does not distinguish different versions of proteins or "proteoforms" derivatized by post translational modifications [1], while top-down proteomic measurements readily distinguish such proteoforms [2]. Both bottom-up and top-down proteomics approaches have distinctive technology challenges associated with extending proteomic applications. This short review discusses bottom-up proteomics sensitivity and top-down proteomic separation techniques, two significant challenges in proteomic technology/methodology that at present provide significant limitations to their applications.
Large-Scale High-Throughput Bottom-Up Proteomics of Single Cells and Small Cell Populations
Bottom-up proteomics has become a largely mature tool for proteome characterization. It is now practical to confidently identify thousands of proteins in a single liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis without the need for sample pre-fractionation and multiple analyses [3-5]. However, bottom-up proteomic analysis presently requires sample sizes ranging from micrograms to high level nanograms (e.g., >100 ng), which largely excludes use of proteomics to characterize the heterogeneity of a biological system (e.g., within a tissue or among circulating cells). Proteomics for such applications needs sufficient sensitivity to enable broad measurement of proteins from picograms to low nanograms of sample sizes (a single mammalian cell on average contains ~0.5 ng protein). Such sensitive proteomics would likely be achievable only by using a bottom-up approach, as both separation and mass spectral analysis for small peptides are much more efficient than for large proteins.
The sensitivity of bottom-up proteomics depends on both the capability of mass spectrometer platform and the effectiveness of the sample separations. Very narrow column LC-advanced mass spectrometers enable detection of hundreds of tryptic peptides from pictograms of proteomic samples (Figure 1; the paired 'spots' confirm the detected species coming from the proteins in cells cultured with N15/N14 media) [6]. The sensitivity is estimated to be sufficient for detection of ~6000 molecules of a peptide [7], or ~6000 protein molecules if the (trypsin) digestion step during sample preparation is quantitative. Such performance, however, has not been to date achievable for tandem mass spectrometry dissociation/sequencing of peptides. Our recent evaluation of a Thermo Orbitrap-Velos tandem mass spectrometer, for example, has been found to enable assignment of >12,000 peptides (at 1% false discovery rate at peptide level, FDR-peptide) and >3000 proteins from 10 ng of a human (MCF- 7) cell lysate tryptic digest (Figure 2), and >4000 peptides (at 1% FDR-peptide) and >1000 proteins from 1 ng microbial Shewanella oneidansis (~5000 open reading frames, ORFs) cell lysate tryptic digest (not shown), representing the state-of-the-art for the sensitivity of tandem mass spectrometry proteomics analysis. Such sensitivity could support, e.g., proteomics investigations of heterogeneity, in samples as small as 20-30 cells (e.g., ~0.4 ng proteins per cancer cells). Very narrow LC columns (20 μm i.d.) are typically required for this ultrasensitive analysis (ESI-MS sensitivity is roughly proportional to the inverse of the square of the LC column diameter due to the effect of flow rate upon sensitivity; Ref. 7), and experimental challenges for use of such narrow LC columns have been effectively addressed [8]. Other formats of LC and capillary zone electrophoresis (CZE) have been recently reported for sensitive bottom-up proteomics; however, the resultant sensitivity and analysis coverage are significantly lower than that obtained by using of narrow packed capillaries described above. For example, CZE enables assignment of only 456 peptides (at 1% FDR-peptide) and 140 proteins from 2 ng of microbial E. coli (~4000 ORFs) lysate tryptic digest [9], while porous layer open tubular column LC has been mainly practiced for microproteomics (i.e., the sample sizes at low level of micrograms) [10].
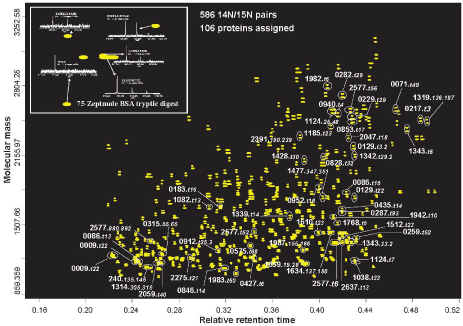
Figure 1 : LC-MS of peptides detected from 0.05-ng D. radiodurans cell lysate tryptic digest. Conditions: 15 μm i.d. x 85 cm column packed with 3 μm C18 is used for LC separation and FTICR mass spectrometer for detection. N15 labeling technique is used for confirmation of the detection for cellular peptides, instead of noise. The LC elution time (0-180 min) is normalized for peptide assignment by accurate mass and time tags approach. The inserted mass spectra show the experiment detection limit examined using BSA tryptic digest (Ref. 6). Experimental details are given in Ref. 7.
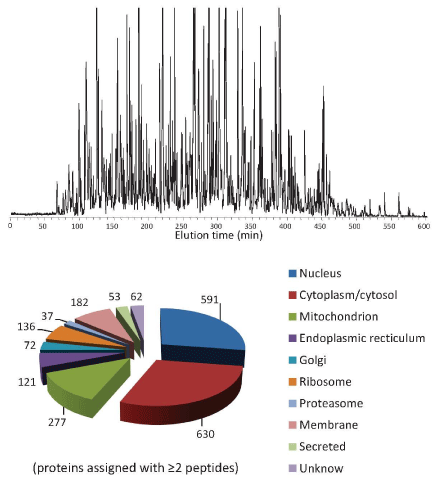
Figure 2 : Ultrasensitive bottom-up proteomics for analysis of 10-ng MCF7 lysate tryptic digest. In a single LC-MS/MS run, 12,458 peptides are assigned (at 1% FDR-peptide) from 3003 proteins. Conditions: 20 μm i.d. x 50 cm column packed with 3 μm C18 for separation with mobile phase gradient from solvent a (100% H2O/0.1% formic acid FA) to 65%B (100% acetonitrile ACN/0.1% FA) in 600 min, Velos Orbitrap CID MS/MS for peptide sequencing, MSGF+ [17] for peptide assignment with control of FDR at peptide level.
Single-cell "discovery mode" proteomics using tandem mass spectrometry would require 10- to 20-fold higher sensitivity than that shown in Figure 2. MS instrumentation continues to rapidly improve in both sensitivity and speed for peptide sequencing [5]. Rapid MS/MS data acquisitions on advanced mass spectrometers allow for use of fast LC to sharpen peptide peaks and improve MS/ MS sensitivity, while continuously narrowing LC columns (e.g., from 15-20 to 5-10 μm i.d.) can further significantly increase LC-MS sensitivity achievable. Integration of these cutting-edge LC-MS platforms and techniques provides a potential base for large-scale single-cell proteomics. However, for this promise to be realized, the "up-front" challenges associated with high-through processing of individual cells, including cell lysis, disposal of cell debris, lysate cleanup, reduction, and digestion remain to be effectively addressed.
Development of High-Efficiency Capillary Separation Techniques for Top-Down Proteomics
Top-down proteomics involves MS analyses that avoid protein digestion, but typically require dissociation of proteins in the MS to distinguish proteoforms that may be crucial to a range of biological processes. It is possible now to sequence proteins and large polypeptides (MWs >100 kDa) on advanced mass spectrometers [2], however, quality MS/MS spectra are much more difficult to obtain for large proteins/polypeptides than for small tryptic peptides, which is a major factor responsible for the low analytical sensitivity of top-down proteomics. Key to improvements will be achieving more effective separations to allow more proteoforms to be identified or distinguished with improved sensitivity.
The major challenge for protein separations arises from the strong interactions between proteins/large polypeptides and separation media, which influence both separation efficiency and sample recovery. Capillary electrophoresis has a significant potential for overcoming this difficulty as the separations are achieved without need for interactions with the separation media. CZE has been explored for tandem mass spectrometry top-down proteomics, e.g., to assign 30-80 kDa proteins by improving sample loading capability [11]. Compared with CZE, LC provide much higher resolution (e.g., with peak capacities of >400) for separation of complex proteomic proteins/polypeptides having MWs of up to >40 kDa [12]. Such high-resolution LC of proteins and polypeptides is achieved by use of weak interactions between proteins/polypeptides and the stationary phases, which exploits the role of LC kinetics in separation of proteins/polypeptides, allows for utilization of long columns and small particle media to improve separation efficiency, as well as improves sample recovery. Short alkyl (e.g., C1-C4) bonded reverse phase packings have been explored for this purpose [12], and the resultant separation efficiency and sensitivity for proteins and large polypeptides are improved over strong interaction separation media (e.g., C8, C8 like or even C18) [2,13-15]. Ultrahigh pressures are particularly useful for delivering the mobile phases through small particles-packed long columns and Figure 3 show a 30K psi RPLC separation that we recently obtained for a cell lysate using a 2-m long column packed with 3-μm particles. The mass spectra inserted show existence of various levels of low abundance proteins and polypeptides (e.g., having MWs from 7.4 to 20 kDa) in a whole cell lysate, which challenges the subsequent tandem mass spectrometry sensitivity and spectral interpretation (e.g., due to low quality isotopic envelopes), especially for large proteins [12]. An automated 40K psi ultrahigh-pressure LC system is now being developed and evaluated in our laboratory for further improvement of LC separation efficiencies and mass spectrometry detection sensitivity, and thus broad use in the characterization of proteoforms.
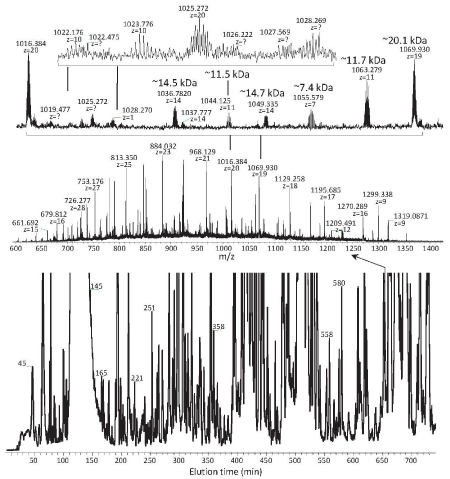
Figure 3 : High-resolution LC-mass spectrometry of proteomic proteins/ polypeptides. Conditions: 2 m L x 100 μm i.d. column packed with 3-μm porous (300 Å) C4-particles for separation of a 2.5-μg of S. Oneideisis cell lysate; mobile phase gradient from solvent A (5% ACN/95% H2O, 0.1% FA) to 80%B (ACN, 0.1% TFA) in 1000 min and Thermo Exactive mass spectrometry for detection.
High-resolution capillary separation techniques have to date been largely unexplored for tandem mass spectrometry top-down proteomics. Various modes of capillary chromatography and electrophoresis based on hydrophilic/hydrophobic interactions, size exclusion, ion exchange, isoelectric focusing (CIEF) etc., need to be carefully re-examined and further improved for their efficiency and sample recovery. Of these capillary separation techniques, CIEF is particularly interesting as it has been proven capable of providing high resolutions for complex proteomic proteins and polypeptides with peak capacities of ~1000 [16] and its power for tandem mass spectrometry characterization of complex mixtures of large proteins (e.g., MWs>50 kDa) needs to be investigated. Also, fractionation and enrichment of proteins and polypeptides by capillary chromatography and electrophoresis need optimized for sensitive, large-scale top-down proteomics.
Acknowledgement
Portions of this research were supported by Department of Energy Office of Biological and Environmental Research Genome Sciences Program under the Pan-Omics project. Experimental work was performed in the Environmental Molecular Sciences Laboratory, a DOE/BER national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, Washington. PNNL is operated by Battelle for the DOE under contract DE-AC05- 76RLO-1830.
References
- Zhang Y, Fonslow BR, Shan B, Baek MC, Yates JR 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem Rev. 2013; 113: 2343-2394.
- Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011; 480: 254-258.
- Shen Y, Zhang R, Moore RJ, Kim J, Metz TO, Hixson KK, et al. Automated 20 kpsi RPLC-MS and MS/MS with chromatographic peak capacities of 1000-1500 and capabilities in proteomics and metabolomics. Anal Chem. 2005; 77: 3090-3100.
- Thakur SS, Geiger T, Chatterjee B, Bandilla P, Fröhlich F, Cox J, et al. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol Cell Proteomics. 2011; 10: M110.
- Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, et al. The one hour yeast proteome. Mol Cell Proteomics. 2014; 13: 339-347.
- Shen Y, ToliÄ N, Masselon C, Pasa-ToliÄ L, Camp DG 2nd, Lipton MS, Anderson GA. Nanoscale proteomics. Anal Bioanal Chem. 2004; 378: 1037-1045.
- Shen Y, Zhao R, Berger SJ, Anderson GA, Rodriguez N, Smith RD. High-efficiency nanoscale liquid chromatography coupled on-line with mass spectrometry using nanoelectrospray ionization for proteomics. Anal Chem. 2002; 74: 4235-4249.
- Shen Y, Moore RJ, Zhao R, Blonder J, Berger SJ, Auberry DL, et al. High-efficiency on-line solid-phase extraction coupled to 15-150-μm-i.d. column liquid chromatography for proteomic analysis. Anal Chem 2003; 75: 3596-3605.
- Zhu G, Sun L, Yan X, Dovichi NJ. Bottom-up proteomics of Escherichia coli using dynamic pH junction preconcentration and capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry. Anal Chem. 2014; 86: 6331-6336.
- Thakur D, Rejta T, Wang D, Bones J, Cha S, Clodfelder-Miller B, et al. Microproteomic analysis of 10,000 laser captured microdissected breast tumor cells using short-range sodium dodecyl sulfate-polyacrylamide gel electrophoresis and porous layer open tubular liquid chromatography tandem mass spectrometry. J Chromatogr A. 2011; 1218: 8168-8174.
- Li Y, Compton PD, Tran JC, Ntai I, Kelleher NL. Optimizing capillary electrophoresis for top-down proteomics of 30-80 kDa proteins. Proteomics. 2014; 14: 1158-1164.
- Shen Y, Tolic N, Zhao R, Shukla AK, Piehowski PD, Kim S, et al. High-resolution reverse-phase columns for UPLC-mass spectrometry top-down proteomics. Anal Chem, submitted.
- Roth MJ, Plymire DA, Chang AN, Kim J, Maresh EM, Larson SE, et al. Sensitive and reproducible intact mass analysis of complex protein mixtures with superficially porous capillary reversed-phase liquid chromatography mass spectrometry. Anal Chem 2011; 83: 9586-9592.
- Tipton JD, Tran JC, Catherman AD, Ahlf DR, Durbin KR, Lee JE, et al. Nano-LC FT-ICR tandem mass spectrometry for top-down proteomics: routine baseline unit mass resolution of whole cell lysate proteins up to 72 kDa. Anal Chem 2011; 84: 2111-2117.
- Wu Z, Wei B, Zhang X, Wirth MJ. Efficient separations of intact proteins using slip-flow with nano-liquid chromatography-mass spectrometry. Anal Chem. 2014; 86: 1592-1598.
- Shen Y, Xiang F, Veenstra TD, Fung EN, Smith RD . High-resolution capillary isoelectric focusing of complex protein mixtures from lysates of microorganisms. Anal Chem. 1999; 71: 5348-5353.
- Kim S, Gupta N, Bandeira N, Pevzner PA. Spectral dictionaries: Integrating de novo peptide sequencing with database search of tandem mass spectra. Mol Cell Proteomics. 2009; 8: 53-69.