
Review Article
Austin J Radiol. 2021; 8(2): 1125.
A Molecular Dynamics of Radiation-Induced DNA Damage Response: Exploring the Pathways of Signaling, Repair, and Cell Death
Singh V¹, Kumar R² and Gautam HK¹*
¹CSIR-Institute of Genomics and Integrative Biology, India
²Radiation Biotechnology Lab, Institute of Nuclear Medicine & Allied Sciences, India
*Corresponding author: Hemant K Gautam, CSIRInstitute of Genomics and Integrative Biology, Sukhdev Vihar, Mathura Road, New Delhi-110025, India
Received: January 25, 2021; Accepted: February 19, 2021; Published: February 26, 2021
Abstract
Purpose: In this review, we summarized the latest information related to accidentally and/or un-accidental exposure of ionizing radiation triggered by oxidative stress and/or cytotoxicity and adverse effects on human health such as hematopoietic, gastrointestinal and cerebrovascular injury collectively referred to as acute radiation syndromes. Directly or indirectly IR induced oxidation of biomolecules, especially DNA, resulting in altered genomic stability and DNA strand breaks. DNA strand breaks are recognized by DNA damage sensory protein that activates downstream checkpoint kinases as well as initiate compensatory multiple intracellular and intranuclear signaling pathways, resulting in cell cycle arrest and DNA repair. Simultaneously activates tumor suppressor genes leading to death signaling pathway or triggering of numerous autocrine/paracrine loops leading to structural dis-organization and programmed cell death. These signaling pathways work together to decrease the magnitude of radiotherapy and promote the development of radiation resistance in cancer cells. The fate of the cells and DNA damage repair depending on the severity of radiation exposure and types of DNA damage.
Conclusions: Based on the recent invested reports related to IR and DNA damage signaling, this review would be helpful for researchers and healthcare providers to develop a new research concept and translate this information into a cancer therapeutic approach. Moreover, target specific screening and development of radiation countermeasures agent for radiological emergencies.
Keywords: IR; Genotoxicity; Cellular Stress; Signaling; DNA repair; Cell survival; Cell death
Introduction
When exposed to ionizing radiation during radiological release or nuclear detonation incident, an act of terrorism, radioactive contamination in public places, unconscious handling of radioactive sources, cause early and late harmful effects on human. Ionizing radiation has sufficient energy to release electrons from atoms or molecules thereby ionize them [1]. It can be allocated into Low LET and High LET (based on relative biological effectiveness), or into weakly penetrating radiation and strongly penetrating radiation (based on ability to penetrate shielding or the human body). High LET emissions includes typically protons, neutrons, and alpha particles (particles of same or high mass), which having ICRP recommends a radiation weighting factor higher than one. In contrast, Low LET radiations typically include photons (χ-rays and γ-rays), electrons, positrons, and muons, which having ICRP recommends a radiation weighting factor equals to one. Most of the radiation sources emit both types of radiation including high and low LET radiation. Low LET radiation deposit less energy and causing less destruction per radiation track as compared to High-LET radiation, [1-3]. The significant effect of radiation-induced death rate is dependent on the quantity and quality of radiation, exposure time, and also the sensitivity of cells and organ systems [4-7]. The danger of irradiation represents different levels of radiation-induced tissue toxicity such as hematopoietic (2-6 Gy), gastrointestinal (6-8 Gy), and cerebrovascular (>8Gy) collectively called acute radiation syndromes [5,8]. To date, there are minimal information and parameters investigated related to characteristic and pathognomonic physical findings at an early stage of radiation exposure. Therefore, there is an urgent need to develop a basic understanding and diagnostic assays to identify at first effects of radiation consequences to minimize the lethal effects of ionizing radiation timely. Moreover, there are so many radiation countermeasures agents that have been developed, and some under in clinical trials [9-13]. However, this problem still unresolved to the medical management of ionizing radiation-induced lethality in a mass casualty scenario. Currently, we use ionizing radiation (as a primary cancer treatment approach (in fractionated doses) because it inhibits cancer cell progression and shrinks tumor size by inducing cytotoxicity mainly disruption of genomic stability (DNA damage) and has considerably controlled the progression of tumor and improved survival of cancer patients. In some cases, recurrence and refractory problems are observed due to the development of radio-resistance and the presence of residual disease after therapy. Multiple factors are involved in recurrence, refractory and radioresistance problem including activation of pro-survival signaling, such as MAPK, AKT, ERK, ATM/ATR, DNA-PKcs, and NF-κB which can suppressed cell death machinery, induced cell cycle arrest, initiate DNA repair mechanisms, cell survival and cell proliferation [14-16]. These signaling pathways cumulatively reduce the degree of radiation-induced cytotoxicity and induce the development of radio-resistance in cancer cells. Hence, selectively targeting these prosurvival signaling pathways has excellent potential to modulate the harmful consequences of ionizing radiation exposure at the cellular, tissue, and organism levels and simultaneously radio-sensitization of cancer cells.
In this review, we focused on understanding the consequences of ionizing radiation a time and dose-dependent manner on various organs of the human system, especially effects on DNA at the molecular level. Based on the available literature, we also summarize the current information on how these radiations and/or oxidative and genotoxic stress-induced activation of intracellular and intranuclear signaling pathways and possible crosstalk relation between them. Moreover, how these signaling pathways play a central role in cell cycle arrest and DNA repair mechanism in ionizing radiationinduced tissue injury and overcome radio-resistance in cancer cells using pro-survival signaling inhibitors.
Ionizing Radiation and Human Health
Ionizing radiation has sufficient energy to damage biological systems primarily due to the macro-molecule lesion (damaged to DNA, lipid, and proteins), which may be the result of direct contact of radiation with macro-molecules and/or indirect interaction by reactive nitrogen and oxygen species, amplified by cellular oxygen. The immediate effect on cells refers to the direct deposition and distribution of radiation energy into a highly sensitive atom or bio-molecule in a cell. Whereas, indirect impact on cell includes absorption of energy by the external medium (water), leading to the production of diffusive intermediates (unstable hyper-oxide molecules) which then attack the sensitive molecules and afflict subcellular structures [17,18]. Certain molecular changes are so complex that it may be tough for the body’s repair mechanisms to restore them correctly. However, the mark is that only a small fraction of such changes would be probable to result in cancer or other health effects [3].
The sensitivity of exposed cells also determines the types of cells and extent of damage; rapidly dividing cells being vulnerable to radiation and differentiated cells (like neurons), muscle, bone, and collagen-producing cells and cancer cells comparatively showed the least consequences of effects of ionizing radiation [4,7]. Exposure of ionizing radiation to humans deposits energy into human tissue, thereby disturbing the healthy anatomic structure and the physiological functions of various organs causes serious public health problems (Figure 1). The most radiation-sensitive organs in the human body include the gastrointestinal, hematopoietic spermatogenic, skin, and vascular systems [17,19-24]. Radiation-induced lethality may be due to local exposure of the body, leading to Local Radiation Injury (LRI) and/or whole-body exposure, leading to Acute Radiation Syndrome (ARS). LRI is generally not life-threatening and includes clinical effects like hair loss, erythema followed by hyperpigmentation, and skin radio-necrosis [25]. Human acute radiation syndrome also called radiation sickness is a severe illness caused by the deposit of IR or internalized radio-nuclides to most or whole body in a relatively short period. Generally, penetrating of high doses of IR causes ARS [8]. ARS comprises penetrating acute radiation doses >1Gy of whole-body radiation exposure or significant partial-body radiation exposure. Sequentially, the main clinical components of ARS include the hematopoietic (2-6 Gy), gastrointestinal (6-8 Gy), and cerebrovascular (>8Gy) sub-syndromes [5].
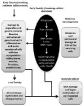
Figure 1: Differential levels of ionizing radiation-induced tissue toxicity.
Early-onset adverse health effects of hematopoietic injury comprise vomiting, nausea, headache, fever fatigue, and temporary skin redness () and later on decline lymphocytes, neutrophils, and platelet counts, hemorrhage collectively(increased susceptibility to infection over some time of radiation exposure [4]. Patients exposed with ionizing radiation doses between 0.2-2 Gy cause transitory arrest in cell cycle and clinically insignificant decline in cell counts but in some cases, mild symptoms such as nausea or headache were seen at 0.35Gy exposure. Absorbed ionizing radiation doses more than 2Gy are produce clinical symptoms include in ARS [26]. Deposition of a high dose of ionizing radiation causes infection and/or hemorrhage and sometimes without significant supportive care, almost half of the people exposed with 3.5Gy will die within 60 days [27,28].
At doses between 6-10 Gy, adverse health effects are seen in Gastrointestinal (GI) tissues along with the hematological injury. The vulnerability and sensitivity of the intestinal tissue to ionizing radiation are due to the fast cell renewal system and proliferating cell compartment of the intestinal crypt and/or villi [29]. The primary symptoms may comprise early nausea, vomiting (rarely severe), anorexia, crampy pain in the abdomen and watery diarrhea are significant symptoms that often occurred within one to two hours post ionizing radiation exposure [30]. Later on, an illness may be manifest, and the patient may experience severe diarrhea with or without fever and vomiting. Moreover, GI syndrome constitutes absorption of abnormal food nutrients, significant imbalance of fluid and electrolyte, GI bleeding and sepsis due to disrupting the integrity of the villus lining causes overwhelming sepsis, renal failure, and possibly cardiovascular collapse. Death from the gastrointestinal injury historically has occurred due to sepsis and complications due to hemorrhage and multisystem organ failure at absorbed doses of 6-10 Gy within 8-14 days post ionizing radiation exposure [27,31].
Neurovascular system and tumor mass show the minimum sensitivity and least consequence of ionizing radiation exposure. The neurovascular syndrome occurs, when people are exposed to a high external dose >10Gy. At these dose levels, clinical features of this syndrome are feeling of burning (just after exposure), nausea and vomiting within minutes, fever, headache and with increasing dose adjust reflexes, hyperpyrexia, prostration, hypotension dizziness, confusion and disorientation, ataxia and unconsciousness [27,31]. All organ systems are severely damaged at this dose, but the damage to the cerebrovascular system is quite severe and usually causes death within 48hrs. Moreover, a lung causes pneumonitis and radiation fibrosis and is mainly due to damage to endothelial cells of small vessels and capillaries [27,32]. Ionizing radiation also induces skin injury, which is manifested in dermal and subcutaneous fibrosis, dry skin with telangiectasias [33]. Unfortunately, peoples exposed to 35Gy and exceed doses damaged large blood vessels and collapsed cardiovascular system, and later on intracranial pressure, cerebral vasculitis, and meningitis may also be seen in most cases. At greater than of 50Gy dose victims will die within two days or less [27,34,35].
In rare cases, clinicians will see a patient with a radiation-induced illness or injury other than an uncommon disease that may present with characteristic findings. Because IR-induced damage usually shows at an early stage of radiation exposure without distinguishing any marks and/or symptoms. There is limited information about early molecular markers and pathognomonic physical findings of ionizing radiation-induced illness. Therefore, this is most promising need to develop a basic understanding and diagnostic tools (biodosimetry and/or biosensors), methods, and assays to diagnose at an early stage of radiation exposure and develop safe and effective mitigators and radiation countermeasure agents to the management of radiation consequences in mass casualty scenario.
Redox Regulation in Cellular Signalling
Low-LET radiation generates large amounts of ROS and RNS (nitric oxide and peroxynitrite) in radiation-exposed mammalian cells. As shown in (Figure 2), ROS are produced mainly by radiolysis of water followed by irradiation (exogenous ROS generation) and leakage of an electron from mitochondrial electron transport chain (endogenous ROS generation) [37]. ROS are short-lived most reactive species include Hydrogen Peroxide (H2O2), superoxide (O2), and Hydroxyl Radicals (OH). On the other hand, RNS, peroxynitrite radical, nitric oxide radical, nitrogen dioxide radical are longer-lived and more specific in their reactions and act to enhance the ROS mediated radiation damage in time and space within the cell. RNS can nitrosylate aromatic amino acid residues, oxidize thiols, damage DNA, and trigger intrinsic mitochondrial apoptotic pathways [36-39]. The overproduction of ROS beyond threshold damaged biomolecules l (mainly DNA, proteins, and lipids) resulting activates intracellular and intranuclear signaling pathways leading to either repair or cell death. Our research group investigated that a small amount of radiation (0.5Gy) that generates ROS is beneficial to alter targeted immunotherapeutic response in hematological malignancies [12,13,40]. In this review, we try to exploit the sequential interplay network of primary consequences of IR such as ROS generation to DNA damage and rapid initial responses of cells, particularly activation of cellular signaling (intracellular and extracellular signaling pathways). These signaling networks play a central role to manage the long-term effects of cell survival from oxidative and genomic instability and maintain cellular homeostasis; regulation of cell survival and cell death.
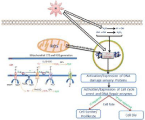
Figure 2: Exogenous and endogenous generation of reactive oxygen
species.
Several signaling pathways are activated in response to ROS levels in the cytoplasm which leads to cell-cycle arrest, mutational status (repairable or not repairable), and induction of cell death [41]. ROS and RNS are observed to inhibit activation of PTPase (protein tyrosine phosphatase) resulting in enhanced tyrosine phosphorylation of multiple proteins such as growth factor receptor family protein [41-43]. It is in the limelight that IR induces only a small fraction of ROS by radiolysis of water and interaction with biomolecules, excess ROS generation is amplified by mitochondria in a Ca2+ -dependent way that can act to inhibit multiple activities of PTPase. IR-induced ROS generation leads to a change in the mitochondrial permeability, which propagates and magnifies the redox signal [44]. All most all type of cell generate ROS and RNS in response to radiation and also initiate activation of receptors tyrosine kinases collectively leads to the promotion of downstream intracellular signaling such as Raf- Ras-MAPK (Mitogen-Activated Protein Kinase) and PI3K/AKT (phosphatidylinositol 3-kinase/AKT or protein kinase B) pathways which maintain cellular status viz cell survival and cell death [45-48]. MAPK pathway regulates diverse processes varying from proliferation and differentiation to apoptosis and includes both pro-survival and pro-apoptotic regulators. JNK and p38 are members of MAPK proapoptotic regulators that promote mitochondrial dysfunction by activating of pro-apoptotic factors like Bax and Bak [49]. MAPK pro-survival regulation includes activation of extracellular signalregulated kinase 1/2 (ERK1/2) which promotes DNA repair and cell growth factors; Jun, Fos through activation of p53 and P21 [50]. Many reports are demonstrated the constitutive activation of Ras increases radio-resistance of cancer cells. In contrast, ERK and MEK activity was attenuated by lovastatin leads to the radio-sensitization of cancer cells [51-53]. In a similar conceptual manner, PI3K-AKT signaling increases the expression of multiple anti-apoptotic proteins such as BCLXL is involved in the radio-resistance of tumor cells. A large number of studies have shown that PI3K-AKT signaling control using pharmacological inhibitors or genetic approaches has increased the radiosensitivity of cancer cells both in vitro and in vivo by reducing DNA repair and inducing programmed cell death [54-56]. In other cell-based models, studies showed that inhibition of PI3K-AKT signalling also involved in increase expression and inactivation of pro-apoptotic markers such as BIM, BAD, and pro-caspases, (Figure 3) [57,58].

Figure 3: Different types of DNA damage followed by ionizing radiation
exposure.
Collectively, based on the above information, IR generates ROS and RNS within the cell and promotes activation of multiple interacting signaling pathways that can either favor or inhibit cell death. Depending on whether pro-apoptotic and anti-apoptotic pathways predominate, the cell will undergo apoptotic/necrotic cell death or will recover from radiation injury.
Oxidative Stress and DNA Damage
An effect of ionizing radiation on DNA is manifested in terms of two indiscriminately destructive processes as described above. As seen in (Figure 4), ionizing radiation-induced disruption in DNA are contributed mainly by Single-Strand Breaks (SSBs) and doublestrand breaks (simple/complex DSBs) with varying complexity such as oxidized base/sugar damage, clustered damage (bistranded and/or tandem), abasic sites and DNA cross-links (Ito et al. 1993). Moreover, depending upon cell type and cell stage-specific responses, low LET radiation-induced lethality causes various types of base lesions such as 450 purine lesions, 850 pyrimidine lesions, 1000 SSBs and 20- 40 DSBs/cell/Gy [59]. Interestingly, during radiotherapy patients exposed with a clinically therapeutic dose around 2Gy/fraction sparsely ionizing radiation causes approximate 3,000 DNA lesions/ exposed cells. This level is far lower as compared to approximately 50,000 lesions formed daily due to ROS in the intracellular milieu [60]. DSBs are more common in radiation-induced damage as compared to SSBs. Phosphodiester bond breaks occur about ten base pairs or less from each other in both strands of the DNA [61,62]. Both simple and complex DSBs have 3’-phosphoglycolate moieties and have single-stranded variable span projections, while complex DSBs have a high level of oxidized base alterations and abasic sites close to the ends of DSBs [63-67]. The number of DSBs rises with the increasing quantity of radiation, starting from a minimal dose of about mGy [68]. Besides, the transcriptionally active DNA is damaged severally and becomes more complex as compared to compact DNA thereby leads to genetic instability, chromosomal alterations, and induction of mutational changes. Thereafter, activate cell-cycle checkpoints, and later on permanent growth arrest or death occurred in affected cells [69,70]. If the checkpoints are inactivated by mutations, the affected cells or tissues showed unwarranted growth culminating in tumor genesis [71].
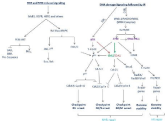
Figure 4: Ionizing radiation-induced oxidative and genotoxic stress and
activation of cellular signaling pathways.
DNA Damage and Nuclear Sensory Signalling Pathways
Cells exposed with a clinically relevant dose of ionizing radiation and any nuclear weapons cause the generation of free radicals and induction of disruption in DNA instability including SSBs, DSBs, clustered damages, structural modification of sugar/base, and also formed DNA-protein cross-links [72]. These DNA modifications are recognized by DNA damage response proteins and trigger the DNA repair process to maintain genome stability cooperatively. The damaged sensory proteins recognized this DNA lesions and recruit DNA repair enzymes at the damage sites. Besides, response/signals are also induced to arrest the cell cycle until the DNA damage is repaired. Some essential proteins accumulate and recognized DNA damage sites across the DNA and initiate the checkpoint kinases activity and cell cycle arrest such as MRN (Mre11/RAD50/Nbs1) complex, DNAPKcs- Ku70/80, PI3K family, ATM (ataxia telangiectasia mutated), ATR (Rad3-related protein) and GFRs in the plasma membrane (e.g., the ERBB family of receptors) as seen in (Figure 3).
The DNA damage related signaling pathways serves as a transduction cascade series for transmitting a signal from DNA damage sensory proteins/receptors to downstream effectors molecules. There are two DNA damage responses (ATM/ATR) signal transduction pathways respond to IR induced DNA damage resulting in induced activation of checkpoint kinases, cell cycle arrest, DNA repair and promotes apoptosis. ATM is one of the vital protein plays a significant role in the signal transduction response to DSBs, and this is found defective in the hereditary disorder ataxia-telangiectasia [73]. Other DNA damage surveillance proteins of this family include Rad3-related protein is response to replication stress [74,75]. ATM and ATR (a member of phosphoinositol 3-kinase like kinase family) and DNA-PKcs are collectively participating in DNA damage response signaling. ATM/ATR cumulatively activates checkpoint kinases that arrest cell cycle progression at G1/S and G2/M transition phases and block entry into mitosis (G2/M), simultaneously promote DNA repair and apoptotic pathways when damage is too severe [76]. The rationale behind slowing down cell cycle progression and takes time to repair damaged sites, thereby correct mutational error and prevent propagation. Several the targets of ATM are tumor suppressor proteins such as p53, Chk2, and H2AX.
These proteins conjointly act as phosphatases and responsible for arresting the cell cycle progression at G1/S or G2/M boundary in healthy cells and regulating cell cycle progression. The cells containing wild-type p53 has ability to control cell cycle progression through inhibiting p21 activity arrest cell cycle in G1 phase, stopping the DNA damage and permitting repair machinery. In contrast, a mutation in p53, despite ionizing radiation-induced DNA damage, will be passed through all cell cycle phases into mitosis. Cell treated with G2 checkpoint kinase inhibitor that exposes the cell to an amplified risk of cytotoxicity by mitotic catastrophe or the transfer of damage to progeny cells. Additionally, p53 can also activate 14-3-3, a protein that results in blocking the G2 phase by sequestering the Cyclin B-Cdk2 complex out of the nucleus. Many pharmacological agents are developed, which block the cell progression through either the G1/S phase or G2/M phase like Β-lapachone, Genistein, Histone deacetylase inhibitors, PcR210 [77]. ATR plays an important role in the homologous recombination repair pathway. Once activated ATR phosphorylates large networks of protein such as Chk1 and Brca1/2 downstream Rad50/51 and DNA repair enzyme PARP1. The overexpression of Rad51 is associated with oncogenic replication stress and tumor progression via genome destabilization [78,79]. Certain cancers harbor homologous recombination defects are successfully treating with PARP1 and Rad51 inhibitors. This strategy shows the successful treatment option for HR-defective (BRCA1/2- mutant) breast and ovarian cancers using Rad 51 and PARP1 inhibitors [80,81].
After detecting a DSBs by sensory proteins ATM and DNAPKcs signaling phosphorylate H2AX (histone variant at serine 139), converting γ-H2AX [82]. After that, phosphorylated H2AX (γ-H2AX) triggers a Chk2signal transduction pathway, subsequent start the functioning of transcription factors p53 and/or Cdc25, resulting in cell cycle stop through the inhibition of cyclins and Cdks activity. ATM/ATR is also directly participating in activating p53, which transcriptionally activates p21, Cdk inhibitor, and prevents cell cycle progression at G1/S boundary [83]. ATM inhibitor not only retarded the activation of DNA-PKcs but also block the recruitment of ku70/80 at DSBs site resulting to enhance radio-sensitivity of cancer cells [84]. ATM role is also manifested in the activation of NF-κB. This transcriptional factor plays a crucial role in cellular immunity l and cell growth through the induction of genetic networks. One more study revealed that ATM controlled transcriptional activation p53 through NF-κB in IR induced genomic alterations [85]. In some cell systems, ATM and DNA-PKcs controlled activation of pro-survival signaling (ERK1/2-and NF-κB) in response to DSBs, which attenuate the apoptotic response following DNA strand breaks [86]. DSBs induced phosphorylation of ATM [82] stimulates phosphorylation of p53. Phosphorylation of p53 induces PIDD activation [87], which then binds with RIP1 (receptor-interacting protein 1) and NEMO (NF-κB -essential modifier, also known as IKKγ) [88]. These molecular events help in the translocation of pATM (phosphorylated ATM) into the nucleus [89] where it phosphorylates NEMO to pNEMO. Thereafter, the complex exits from the nucleus where it binds with the IκB-NF- κB complex and induced activation of IκB kinase. Phosphorylated IκB kinase catalyzes and releases NF-κB from its inhibitor (IκBa or IκBΒ) and translate into the nucleus. The phosphorylated NF-κB assembly translocates from the cytoplasm to the nucleus and regulates its target genes. NF-κB mediated signaling plays an adaptive role in DNA repair, checkpoint regulation, antioxidants level, cell survival, and cell death and also controls the expression of cytokine and chemokine followed by radiation injury [90]. Many pharmacological agents are synthesized, which can stimulate the activation of NF-κB. Cleveland Clinic Foundation has studied the role of the CBLB series of TLR specific agonists, which culminates in NF-κB activation [13].
DNA Repair Mechanism and Cellular Signalling
Human cells have advanced levels of DNA damage repair mechanisms to deal with oxidative stress-induced damage and/ or direct energy deposition. The response to cellular damage can preserve the integrity and stability of the genome to reduce the onset of possible tumor genesis and the aging process. It is faster and quicker to repair a single double-strand break than multiple damaged sites in DNA. The repair of SSBs is usually error-free, but DSBs can be either error-free or error-prone. Some studies suggested that the numerous DSBs followed by low LET radiation exposure would be occurred in 30-60 minutes, while a minute fraction of DSBs, normally <20%, would be less willingly repaired in mammalian cells and some could carry on for >24h [73,84,91-94]. Nevertheless, the repair system, with its genes and proteins is the caretaker of the genome. When cells become deficient in any of the repair proteins, they fail to repair the DNA damage (especially: DSBs) correctly, and this leads to induction of programmed cell death and /or induction of leading to cancer with defective cell cycles regulatory checkpoints (Figure 5).
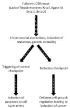
Figure 5: Defects in DNA repair and subsequent defective checkpoint can
lead to cancer induction.
Homologous Recombination (HR) and Non-Homologous End-Joining (NHEJ) are two separate and complementary DSB repair processes that effectively repair activated in the majority of DNA damage [71]. The Base Excision Repair (BER) is the primary mechanism for restoring clustered DNA damage sites in which base lesions are removed near DSBs termini, consistent with the observation that complex DSBs are re-joined before removing base lesions [65,95]. NHEJ and HR pathways are generally called “error-prone” and “error-free” respectively, but mostly this is an oversimplification [96]. While HR provides greater fidelity to repair as compared to NHEJ, the latter is the crucial way to restore prompt DSBs in all cell cycle phases. However, the majority (80-90 %) of DSB repair involves the NHEJ repair pathway [96,97]. On sensing the broken ends that cannot be precisely re-joined, NHEJ directs repair by either deleting or inserting few bases. This repair typically involves restoring “micro homology,” i.e. alignment of one or few complementary bases.
In a mammalian cell, NHEJ is a stepwise response, which is initiated with limited end-processing by MRN complex (Mre11/ RAD50/Nbs1). This is followed by recognition of free DNA ends by Ku proteins and its subsequent binding at DSB [98]. Once bound to DNA ends, the heterodimeric Ku70/Ku80 proteins recruit DNA dependent proteins kinase catalytic subunits (DNA-PKcs) to the DSB termini. The Ku70/80 complex is recruited all most all DSBs, but DNA-PKcs complex only recruited during long-lived DSBs complex [84,99]. This results in the formation of trimeric DNA-PKcs holoenzyme (MRN complex, Ku dimeric subunits & DNA-PKcs). DNA-PKcs component of holoenzyme phosphorylates itself along with other targets including RPA, WRN, and Artemis and polymerase (μ and λ). DNA-PKcs also form γ-H2AX, a phosphorylated product of H2AX in a cell lacking ATM [100,101]. Radiation-induced DSB, i.e. 5’ and 3’ overhangs, hairpins, gaps, flaps, and different loop configurations are trimmed with Artemins and DNA-PKcs endonuclease and DNA polymerase (μ and p). Finally, the break is ligated by DNA ligase IV in association with its binding partners XRCC4 and XLF [102,103].
Fine-tuning of nuclease and polymerase is required for proper ligation activity of Ligase IV. The appropriate functioning of these enzymes with their activation in correct sequence forms the basis of the proper functioning of classical NHEJ repair. An alternative Ligase III mediated NHEJ repair mechanism also exists which acts as an additional contributor in SSB and DSB repair. This repair is facilitated by an abundant nuclear eukaryotic enzyme, Poly (ADP-ribose) polymerase-1 (PARP-1), but it appears to be comparatively more susceptible to error-prone than traditional NHEJ repair [104,105]. PRAP-1 competes with Ku to find broken DNA ends and is followed by ligation by ligase III [105]. ATM is another crucial player that contributes to survival after radiation-induced DNA damage, which repairs a defined subset of DSBs (10%) in the G1 phase in cooperation with Artemis [97,106].
Homologous Recombination (HR) represents another pathway for DSB as well as SSB repair and is an active repair process occurring mainly in the late S phase/G2 phase. HR uses homologous sequences (sister chromatids, repeated regions on the same or different chromosomes, or homologous chromosomes) culminating in the high-fidelity repair of broken ends. This comprises a series of associated sub-pathways that use DNA strand invasion and templatedriven DNA repair synthesis. The homologous recombination repair pathway involved large networks of protein such as ATM, Chk1, and Brca1/2 downstream Rad50/51 and DNA repair enzyme PARP-1. The initial phase (pre-synapsis) of DSB processing consists of attaching of Rad51 filament to a 3’ overhanging tail, IR induced DSBs, which appears to require the complex MRN (Mre11-Rad50-Nbs1) in particular [107,108]. Mre11 is an endo-nuclease that binds directly to DNA, Rad50 and Nbs1 are help in the organizing of MRN complex. Rad50 has ATP-Binding Cassette (ABC) ATPase, Zn hook, and coiled coils that join DSBs and help with Mre11 finishing processing. Thus, Rad50 belongs to the Structural Maintenance of Chromosome (SMC) group of proteins. Nbs1 contributes to the regulatory role of the MRN complex due to its N-terminal phosphopeptide, which assists in the interaction between the C-terminal of ATM, Mre11 subunits, and FHA and BRCT domains. In short, MRN serves as a sensor of DSBs [8]. After recognition of DSBs, cellular machinery tries to find the complementary sequences called homology search. The DNA strand invasion and search for homology are jointly called synapses and are supported by RPA and Rad51, respectively [107]. The invasion of the 3’ end of DNA primes the synthesis of DNA of the DNA duplex template, resulting in intermediate D loop production. Double strand breaks repair occurred at DSB’s second end, either by capturing the second end via DNA annealing or a second invasion event. Generally, second-end annealing is catalyzed using Rad52 protein, which has an exclusive role of annealing complementary ssDNA linked to RPA [109]. Resulting, Double Holiday Junctions (dHJ) are formed which are converted either into non-crossover products by BLM-TOPOIIIa or for resolution into crossover/non-crossover products by a structure-specific endonuclease. The resolvase help in the separation of holiday junctions into crossover and non-crossover products. Inefficient repair of DNA results in genetic instability, which, in turn, can increase the rate of cancer development Indeed, deficiencies in various types of repair pathways are becoming increasingly accepted as fundamental to the etiology of most human tumors.
Bystander Signalling
Ionizing radiation not only affects the cells and cell components but also shows biological effects nearby of the cells. There is plenty of evidence that irradiation can lead to mutation in cells directly or indirectly through nearby irradiated cells. This phenomenon is commonly referred to as the bystander effect by paracrine feedback signaling that may cause carcinogenic effects to normal tissue [110,111]. This is one of the big problems that remain to recurrent tumor relapse following treatment of primary tumor. A large number of studies showed that radiation-induced bystander effects exaggerate the effect of small doses of radiation. Cell to cell communication occurred by gap junction and soluble mediators released by irradiated cells, both collectively play an essential role in the bystander response, it is also stated that the specific signaling pathways are involved [112]. This is noted that the progeny of non-targeted cells shows an increase in genomic instability as demonstrated by the rise in delayed mutations and chromosomal aberrations several generations later indicate the need for a comprehensive evaluation of the bystander problem, especially among genetically susceptible populations. The mechanism of this non-targeted response was studied using in vitro as well as in vivo models. Such studies provide insight on the essence of the signaling molecule(s) that will be invaluable in assessing the clinical significance of the bystander effect and how the bystander phenomenon can be exploited to improve radiotherapy therapeutic benefit. It is well reported that Cyclooxygenase-2 (COX-2) signaling plays an important role in the bystander signaling followed by various growth factors and cytokines such as Transforming Growth Factor β (TGF-β), Tumor Necrosis Factor a (TNF-a), Interleukin 1β (IL1β), and multiple stressors also [112,113]. It is confirmed that IGFBP-3 and COX-2 gene expression is constantly altered in the bystander cells. Signals transmitted through Ras/Raf/MEK/ERK/AP1 cascade reaction and NF-κB pathways, thus finally targeted COX-2 gene transcription and were found three-fold changes in the bystander cells. A specific inhibitor of COX-2; NS-398 neutralizes the effect of the COX-2 signaling pathway in bystander cells. The bystander mutagenic effect in NHLF cells was reduced by 6-fold in the presence of COX-2 inhibitor NS-398. One more selective COX-2 inhibitor, Meloxicam facilitates hematopoietic recovery in sub-lethally irradiated mice and is radiation-protective when given before irradiation [114]. One other gene identified to be expressed in NHLF bystander cells is IGFBP-3, to which the majority of circulating IGFs are bound in bystander cells and prevent them from binding to IGF receptors on the cell surface [115]. Besides, there is evidence that TGFβ in medium transfer studies may play a significant role in mediating bystander effects [116]. It is attributed that the pro-mitogenic reaction of a particle-induced rises the level of transforming growth factor β1 (TGF-β1) in cell supernatants. Cells treated with TGF-β1 containing supernatants induce intracellular ROS generation in untreated cells resulting in decreased levels of TP53 and CDKN1A while CDC2 and Proliferating Nuclear Antigen (PCNA) is increased in the latter. It is well understood that NF-κB and p38 MAPK collectively control COX-2 levels in response to an inflammatory stimulus involving of interleukin (IL)-1β, tumor necrosis factor-a (TNF-a), and interferon-γ (IFN-γ). Nitric oxide also involved in COX-2 mediated bystander effect as nitric oxide and also control expression of IL-8 in human cells [117]. Hence, the study of Bystander effects is one of the crucial aspects for studying mechanisms for radiations induced lethality, and the appearance of COX-2 inhibitors may function to ameliorate the non-targeted cell injury [118-123].
Conclusion and Future Directions
An accident and un-accidentally people exposed to radiation daily. Over the past few years, researchers have been investigated the adverse role of ionizing radiation on human health, especially on DNA at molecular levels. Here, we summarized current information related to the generation of oxidative and genotoxic stress resulting in activation of DNA damage sensory proteins and downstream activate checkpoint kinases as well as initiate compensatory multiple intracellular and intranuclear signaling pathways, resulting prevent cell cycle progression and started DNA repair mechanism. These signaling pathways work together to reduce the extent of radiotherapy and promote the development of radiation resistance in cancer cells. Simultaneously activates tumor suppressor genes leading to death signaling pathway or triggering of numerous autocrine/paracrine loops leading to structural dis-organization and programmed cell death. The fate of the cells and DNA damage repair depending on the severity of radiation exposure and types of DNA damage. Moreover, in this review, we also focused on understanding the role of bystander signaling in tissue injury and repairing in radiological consequences. The primary aim of this article is to understand the consequences of ionizing radiation and stimulate some cutting edge research concepts based on understating the spectrum of DNA damage and repair mechanisms followed by IR exposure. Thus, by selectively targeting these pro-survival pathways, we can mend harmful consequences of ionizing radiation exposure at cellular, tissue, and organism levels and simultaneously radio-sensitize of cancer cells.
Author Contributions
VS wrote the manuscript and final manuscript editing done by HKG, VS, and RK.
Acknowledgement
The authors are grateful to the Indian Council of Medical Research, India for the research associate fellowship award (to VS). We are also thankful to the Director of CSIR-IGIB for providing the opportunity and support to carry out our research work and prepare this manuscript.
References
- Hall EJ, Giaccia AJ. Radiobiology for the radiologist, Lippincott Williams & Wilkins, Philadelphia. 2006b.
- Goodwin PN, Quimby E, Morgan RH, Glasser O. Physical foundations of radiology, Medical Dept. New York. 1970.
- Radiation CAHRELLI, Council NR. Health Risks from Exposure to Low Levels of Ionizing Radiation. BEIR VII Phase 2. National Academies Press. 2006.
- Coleman CN, Stone HB, Moulder JE, Pellmar TC. Medicine. Modulation of radiation injury. Science. 2004; 304: 693-694.
- Hall EJ, Giaccia AJ. Radiobiology for the Radiologist, Lippincott Williams & Wilkins. 2006a.
- Pathak R, Kumar R, Gautam HK. Cross-Species Induction and Enhancement of Antimicrobial Properties in Response to Gamma Irradiation in Exiguobacterium sp. HKG 126. Indian J Microbiol. 2013; 53: 130-136.
- Zajtchuk R, Jenkins D, Walker R, Cerveny T, Alt L, Bogo V, Dons R, Farzaneh N. Textbook of Military Medicine. Part 1. Warfare, Weaponry, and the Casualty. Medical Consequences of Nuclear Warfare. 1989; 2: 300.
- Xiao M, Whitnall MH. Pharmacological countermeasures for the acute radiation syndrome. Curr Mol Pharmacol. 2009; 2: 122-133.
- Arora R, Gupta D, Chawla R, Sagar R, Sharma A, Kumar R, et al. Radioprotection by plant products: present status and future prospects. Phytother Res. 2005; 19: 1-22.
- Dhaker A, Marwah R, Origanti DR, Gupta D, Gautam H, Sultana S, Arora R. In vitro evaluation of antioxidant and radioprotective properties of a novel extremophile from mud volcano: Implications for management of radiation emergencies. Molecular and cellular biochemistry. 2011; 353: 243-250.
- Mishra S, Gupta AK, Malhotra P, Singh P, Pathak R, Kukreti S, et al. Protection Against Ionizing Radiation Induced Oxidative Damage to Structural and Functional Proteins by Semiquinone Glucoside Derivative Isolated from Radioresistant Bacterium Bacillus sp. INM-1. Current Biotechnology. 2014; 3: 117-126.
- Singh S, Kumar R, Malhotra P, Gupta AK, Singh D, Adhikari M. Proteomic & Bioinformatic analysis of molecular mechanisms of radioprotection offered by radioresistant bacterial metabolite RKIP-006. 2018.
- Singh V, Gupta D, Arora R. NF-κB as a key player in regulation of cellular radiation responses and identification of radiation countermeasures. 2015.
- Gupta A, Pathak R, Singh B, Gautam H, Kumar R, Kumar R, et al. Proteomic Analysis of Global Changes in Protein Expression During Exposure of Gamma Radiation in Bacillus sp HKG 112 Isolated from Saline Soil. Journal of microbiology and biotechnology. 2011; 21: 574-581.
- Malhotra P, Gupta AK, Singh D, Mishra S, Singh S, Kumar R. Protection to immune system of mice by N-Acetyl Tryptophan Glucoside (NATG) against gamma radiation induced immune suppression. Molecular immunology. 2019; 114,: 578-590.
- Verma N, Chakrabarti R, Das R, Gautam H. Anti-Inflammatory Effects of Shea Butter through Inhibition of Inos, Cox-2, and Cytokines via the Nf-κb Pathway in Lps-Activated J774 Macrophage Cells. Journal of complementary & integrative medicine. 2012; 9.
- Bomanji JB, Novruzov F, Vinjamuri S. Radiation accidents and their management: emphasis on the role of nuclear medicine professionals. Nucl Med Commun. 2014; 35: 995-1002.
- Kumar R, Gautam H. Understanding the mysterious radioresistance and genomic integrity of Deinococcus radiodurans against gamma radiation: Implications in radiation response modulation in higher organisms. Molecular Biology of Bacteria. 2013; 143-156.
- Fliedner TM, Graessle D, Meineke V, Dorr H. Pathophysiological principles underlying the blood cell concentration responses used to assess the severity of effect after accidental whole-body radiation exposure: an essential basis for an evidence-based clinical triage. Exp Hematol. 2007; 35: 8-16.
- MacNaughton WK. Review article: new insights into the pathogenesis of radiation-induced intestinal dysfunction. Aliment Pharmacol Ther. 2000; 14: 523-528.
- Meistrich ML, Kangasniemi M. Hormone treatment after irradiation stimulates recovery of rat spermatogenesis from surviving spermatogonia. J Androl. 1997; 18: 80-87.
- Moulder J. Post-irradiation approaches to treatment of radiation injuries in the context of radiological terrorism and radiation accidents: A review. International journal of radiation biology. 2004; 80: 3-10.
- Muller K, Meineke V. Radiation-induced alterations in cytokine production by skin cells. Exp Hematol. 2007; 35: 96-104.
- Rodemann HP, Blaese MA. Responses of normal cells to ionizing radiation. Semin Radiat Oncol. 2007; 17: 81-88.
- Arora R, Chawla R, Marwah R, Kumar V, Goel R, Arora P, et al. Medical radiation countermeasures for nuclear and radiological emergencies: Current status and future perspectives. J Pharm Bioallied Sci. 2010; 2: 202- 212.
- Lopez M, Martin M. Medical management of the acute radiation syndrome. Rep Pract Oncol Radiother. 2011; 16: 138-146.
- Institute AFRR, Military US. Medical Management of Radiological Casualties-Fourth Edition, July 2013-Ionizing Radiation and Radionuclide Emergency Treatment, Acute Radiation Syndrome, Skin, Decontamination, Delayed Effects, Independently Published. 2017.
- Waselenko JK, MacVittie TJ, Blakely WF, Pesik N, Wiley AL, Dickerson WE, et al. Medical management of the acute radiation syndrome: recommendations of the Strategic National Stockpile Radiation Working Group. Ann Intern Med. 2004; 140: 1037-1051.
- Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001; 293: 293-297.
- Jarrett DG, Office, A.F.R.R.I.M.M.O. Medical Management of Radiological Casualties Handbook, Armed Forces Radiobiology Research Institute, Military Medical Operations Office. 1999.
- Hall EJ. Radiobiology for the Radiologist, Lippincott Williams & Wilkins. 2000.
- Movsas B, Raffin TA, Epstein AH, Link CJ. Pulmonary radiation injury. Chest. 1997; 111: 1061-1076.
- Barabanova AV. Significance of beta-radiation skin burns in Chernobyl patients for the theory and practice of radiopathology. Vojnosanit Pregl. 2006; 63: 477-480.
- Donnelly EH, Nemhauser JB, Smith JM, Kazzi ZN, Farfan EB, Chang AS, et al. Acute radiation syndrome: assessment and management. South Med J. 2010; 103: 541-546.
- Prasad KN. Handbook of Radiobiology, Taylor & Francis. 1995.
- Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods Enzymol. 1994; 233: 229-240.
- Buchczyk DP, Briviba K, Hartl FU, Sies H. Responses to peroxynitrite in yeast: Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) as a sensitive intracellular target for nitration and enhancement of chaperone expression and ubiquitination. Biol Chem. 2000; 381: 121-126.
- Choi BM, Pae HO, Jang SI, Kim YM, Chung HT. Nitric oxide as a proapoptotic as well as anti-apoptotic modulator. J Biochem Mol Biol. 2002; 35: 116-126.
- Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996; 313: 17-29.
- Singh V, Gupta D, Arora R, Tripathi RP, Almasan A, Macklis RM. Surface levels of CD20 determine anti-CD20 antibodies mediated cell death in vitro. PloS one. 2014; 9: e111113.
- Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003; 22: 5734-5754.
- Galabova-Kovacs G, Kolbus A, Matzen D, Meissl K, Piazzolla D, Rubiolo C, et al. ERK and beyond: insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle. 2006; 5: 1514-1518.
- Tonks NK. Protein tyrosine phosphatases and the control of cellular signaling responses. Adv Pharmacol. 1996; 36: 91-119.
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive Oxygen Species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000; 192: 1001-1014.
- Cui W, Yazlovitskaya EM, Mayo MS, Pelling JC, Persons DL. Cisplatininduced response of c-jun N-terminal kinase 1 and extracellular signal-- regulated protein kinases 1 and 2 in a series of cisplatin-resistant ovarian carcinoma cell lines. Mol Carcinog. 2000; 29: 219-228.
- Dent P, Reardon DB, Park JS, Bowers G, Logsdon C, Valerie K, et al. Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death. Mol Biol Cell. 1999; 10: 2493-2506.
- Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003; 22: 5885-5896.
- McKenna WG, Muschel RJ, Gupta AK, Hahn SM, Bernhard EJ. The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene. 2003; 22: 5866-5875.
- Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006; 281: 21256-21265.
- Astsaturov I, Cohen RB, Harari P. Targeting epidermal growth factor receptor signaling in the treatment of head and neck cancer. Expert Rev Anticancer Ther. 2006; 6: 1179-1193.
- Abbott DW, Holt JT. Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J Biol Chem. 1999; 274: 2732-2742.
- Fritz G, Brachetti C, Kaina B. Lovastatin causes sensitization of HeLa cells to ionizing radiation-induced apoptosis by the abrogation of G2 blockage. Int J Radiat Biol. 2003; 79: 601-610.
- Yan Y, Black CP, Cao PT, Haferbier JL, Kolb RH, Spieker RS, et al. Gammairradiation- induced DNA damage checkpoint activation involves feedback regulation between extracellular signal-regulated kinase 1/2 and BRCA1. Cancer Res. 2008; 68: 5113-5121.
- Kim IA, Bae SS, Fernandes A, Wu J, Muschel RJ, McKenna WG, et al. Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res. 2005; 65: 7902-7910.
- Shimura T, Kakuda S, Ochiai Y, Kuwahara Y, Takai Y, Fukumoto M. Targeting the AKT/GSK3beta/cyclin D1/Cdk4 survival signaling pathway for eradication of tumor radioresistance acquired by fractionated radiotherapy. Int J Radiat Oncol Biol Phys. 2011; 80: 540-548.
- Toulany M, Kehlbach R, Florczak U, Sak A, Wang S, Chen J, et al. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNAPKcs- dependent DNA double-strand break repair. Mol Cancer Ther. 2008; 7: 1772-1781.
- Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez- Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004; 30: 193-204.
- Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem. 2007; 282: 21206-21212.
- Cadet J, Douki T, Ravanat JL. Oxidatively generated damage to the guanine moiety of DNA: mechanistic aspects and formation in cells. Acc Chem Res. 2008; 41: 1075-1083.
- Lomax ME, Folkes LK, O’Neill P. Biological consequences of radiationinduced DNA damage: relevance to radiotherapy. Clin Oncol (R Coll Radiol). 2013; 25: 578-585.
- Hanai R, Yazu M, Hieda K. On the experimental distinction between ssbs and dsbs in circular DNA. Int J Radiat Biol. 1998; 73: 475-479.
- Van Der Schans GP. Gamma-ray induced double-strand breaks in DNA resulting from randomly-inflicted single-strand breaks: temporal local denaturation, a new radiation phenomenon? Int J Radiat Biol Relat Stud Phys Chem Med. 1978; 33: 105-120.
- Datta K, Jaruga P, Dizdaroglu M, Neumann RD, Winters TA. Molecular analysis of base damage clustering associated with a site-specific radiationinduced DNA double-strand break. Radiat Res. 2006; 166: 767-781.
- Datta K, Neumann RD, Winters TA. Characterization of complex apurinic/ apyrimidinic-site clustering associated with an authentic site-specific radiation-induced DNA double-strand break. Proc Natl Acad Sci USA. 2005; 102: 10569-10574.
- Datta K, Purkayastha S, Neumann RD, Pastwa E, Winters TA. Base damage immediately upstream from double-strand break ends is a more severe impediment to nonhomologous end joining than blocked 3’-termini. Radiat Res. 2011; 175: 97-112.
- Henner WD, Grunberg SM, Haseltine WA. Sites and structure of gamma radiation-induced DNA strand breaks. J Biol Chem. 1982; 257: 11750- 11754.
- Henner WD, Rodriguez LO, Hecht SM, Haseltine WA. Gamma Ray induced deoxyribonucleic acid strand breaks. 3’ Glycolate termini. J Biol Chem. 1983; 258: 711-713.
- Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 2003; 100: 5057-5062.
- Claesson K, Magnander K, Kahu H, Lindegren S, Hultborn R, Elmroth K. RBE of alpha-particles from (211). At for complex DNA damage and cell survival in relation to cell cycle position. Int J Radiat Biol. 2011; 87: 372-384.
- Magnander K, Hultborn R, Claesson K, Elmroth K. Clustered DNA damage in irradiated human diploid fibroblasts: influence of chromatin organization. Radiat Res. 2010; 173: 272-282.
- Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001; 27: 247-254.
- Bourguignon MH, Gisone PA, Perez MR, Michelin S, Dubner D, Giorgio MD, et al. Genetic and epigenetic features in radiation sensitivity Part I: cell signalling in radiation response. Eur J Nucl Med Mol Imaging. 2005; 32: 229- 246.
- Jeggo PA, Lobrich M. Artemis links ATM to double strand break rejoining. Cell Cycle. 2005; 4: 359-362.
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010; 108: 73-112.
- Yazinski SA, Zou L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu Rev Genet. 2016; 50: 155-173.
- Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA doublestrand break repair during the mammalian cell cycle. Mol Cell Biol. 2003; 23: 5706-5715.
- Dumont F, Le Roux A, Bischoff P. Radiation countermeasure agents: an update. Expert Opin Ther Pat. 2010; 20: 73-101.
- Nagathihalli NS, Nagaraju G. RAD51 as a potential biomarker and therapeutic target for pancreatic cancer. Biochim Biophys Acta. 2011; 1816: 209-218.
- Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004; 23: 546-553.
- Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011; 5: 387-393.
- Livraghi L, Garber JE. PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med. 2015; 13: 188.
- Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATMdependent signaling pathways. DNA Repair (Amst). 2004; 3: 889-900.
- Kimura SH, Nojima H. Cyclin G1 associates with MDM2 and regulates accumulation and degradation of p53 protein. Genes Cells. 2002; 7: 869- 880.
- Reynolds P, Anderson JA, Harper JV, Hill MA, Botchway SW, Parker AW, et al. The dynamics of Ku70/80 and DNA-PKcs at DSBs induced by ionizing radiation is dependent on the complexity of damage. Nucleic Acids Res. 2012; 40: 10821-10831.
- Rashi-Elkeles S, Elkon R, Weizman N, Linhart C, Amariglio N, Sternberg G, et al. Parallel induction of ATM-dependent pro- and antiapoptotic signals in response to ionizing radiation in murine lymphoid tissue. Oncogene. 2006; 25: 1584-1592.
- Panta GR, Kaur S, Cavin LG, Cortes ML, Mercurio F, Lothstein L, et al. ATM and the catalytic subunit of DNA-dependent protein kinase activate NFkappaB through a common MEK/extracellular signal-regulated kinase/p90 (rsk) signaling pathway in response to distinct forms of DNA damage. Mol Cell Biol. 2004; 24: 1823-1835.
- Lin Y, Ma W, Benchimol S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet. 2000; 26: 122-127.
- May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000; 289: 1550-1554.
- Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell. 2005; 123: 1079-1092.
- Ahmed KM, Li JJ. NF-kappa B-mediated adaptive resistance to ionizing radiation. Free Radic Biol Med. 2008; 44: 1-13.
- Asaithamby A, Uematsu N, Chatterjee A, Story MD, Burma S, Chen DJ. Repair of HZE-particle-induced DNA double-strand breaks in normal human fibroblasts. Radiat Res. 2008; 169: 437-446.
- Botchway SW, Stevens DL, Hill MA, Jenner TJ, O’Neill P. Induction and Rejoining of DNA Double-Strand Breaks in Chinese Hamster V79-4 Cells Irradiated with Characteristic Aluminum K and Copper L Ultrasoft X Rays. Radiation Research. 1997; 148: 317-324.
- Jakob B, Splinter J, Conrad S, Voss KO, Zink D, Durante M, et al. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011; 39: 6489-6499.
- Schmid TE, Dollinger G, Beisker W, Hable V, Greubel C, Auer S, et al. Differences in the kinetics of gamma-H2AX fluorescence decay after exposure to low and high LET radiation. Int J Radiat Biol. 2010; 86: 682-691.
- Dobbs T, Palmer P, Maniou Z, Lomax M, O’Neill P. Interplay of two major repair pathways in the processing of complex double-strand DNA breaks. DNA repair. 2008; 7: 1372-1383.
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008; 18: 134-147.
- Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004; 16: 715-724.
- Jeggo P, Lobrich M. Radiation-induced DNA damage responses. Radiat Prot Dosimetry. 2006; 122: 124-127.
- Peddi P, Francisco DC, Cecil AM, Hair JM, Panayiotidis MI, Georgakilas AG. Processing of clustered DNA damage in human breast cancer cells MCF-7 with partial DNA-PKcs deficiency. Cancer Lett. 2008; 269: 174-183.
- Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNAPK. Oncogene. 2005; 24: 949-961.
- Stucki M, Jackson SP. GammaH2AX and MDC1: anchoring the DNAdamage- response machinery to broken chromosomes. DNA Repair (Amst). 2006; 5: 534-543.
- Dai Y, Kysela B, Hanakahi LA, Manolis K, Riballo E, Stumm M, et al. Nonhomologous end joining and V(D)J recombination require an additional factor. Proc Natl Acad Sci USA. 2003; 100: 2462-2467.
- Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res. 2011; 711: 61-72.
- Audebert M, Salles B, Calsou P. Involvement of poly (ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004; 279: 55117-55126.
- Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006; 34: 6170-6182.
- Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009; 28: 3413-3427.
- Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008; 18: 99-113.
- Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007; 85: 509-520.
- Sugiyama T, Kantake N, Wu Y, Kowalczykowski SC. Rad52-mediated DNA annealing after Rad51-mediated DNA strand exchange promotes second ssDNA capture. EMBO J. 2006; 25: 5539-5548.
- Clutton SM, Townsend KM, Walker C, Ansell JD, Wright EG. Radiationinduced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis. 1996; 17: 1633-1639.
- Nagasawa H, Cremesti A, Kolesnick R, Fuks Z, Little JB. Involvement of Membrane Signaling in the Bystander Effect in Irradiated Cells. Cancer Research. 2002; 62: 2531-2534.
- Zhou H, Ivanov VN, Gillespie J, Geard CR, Amundson SA, Brenner DJ, et al. Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc Natl Acad Sci USA. 2005; 102: 14641-14646.
- Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA. 1993; 90: 11693- 11697.
- Hofer M, Pospisil M, Dusek L, Hoferova Z, Weiterova L. A single dose of an inhibitor of cyclooxygenase 2, meloxicam, administered shortly after irradiation increases survival of lethally irradiated mice. Radiat Res. 2011; 176: 269-272.
- Grimberg A. P53 and IGFBP-3: apoptosis and cancer protection. Mol Genet Metab. 2000; 70: 85-98.
- Iyer R, Lehnert BE, Svensson R. Factors underlying the cell growth-related bystander responses to alpha particles. Cancer Res. 2000; 60: 1290-1298.
- Singer CA, Baker KJ, McCaffrey A, AuCoin DP, Dechert MA, Gerthoffer WT. p38 MAPK and NF-kappaB mediate COX-2 expression in human airway myocytes. Am J Physiol Lung Cell Mol Physiol. 2003; 285: L1087-L1098.
- Crofford LJ, Lipsky PE, Brooks P, Abramson SB, Simon LS, van de Putte LB. Basic biology and clinical application of specific cyclooxygenase-2 inhibitors. Arthritis Rheum. 2000; 43: 4-13.
- Medical Management of Radiological Casualties, Second Edition, Handbook, armed forces radiobiology research instbethesda md. 2003.
- Ito T, Baker SC, Stickley CD, Peak JG, Peak MJ. Dependence of the yield of strand breaks induced by gamma-rays in DNA on the physical conditions of exposure: water content and temperature. Int J Radiat Biol. 1993; 63: 289-296.
- Lobrich M, Jeggo PA. Harmonising the response to DSBs: a new string in the ATM bow. DNA Repair (Amst). 2005; 4: 749-759.
- Singh V. Differential Action of Anti-CD20 Monoclonal Antibodies: Role in Induction of Cell Death. Journal of Cancer Science and Therapy. 2018; 10: 064-068.
- Singh V, Gupta D, Almasan A. Development of novel anti-CD20 monoclonal antibodies and modulation in CD20 levels on cell surface: looking to improve immunotherapy response. Journal of cancer science & therapy. 2015; 7: 347-358.