
Case Report
Austin J Reprod Med Infertil. 2015;2(1): 1004.
Unicornuate Uterus with Multiple Skeletal Defects and Large Regions of Homozygosity
Dania Al-Jaroudi*, Suha Tashkandi, Mamoun Al-Awad, Faryal Khan
Department of Obstetrics and Genecology, Reproductive Endocrinology and Infertility Medicine Department, Women’s Specialized Hospital, Riyadh, Saudi Arabia
*Corresponding author: Dania Al-Jaroudi, Department of Obstetrics and Gynecology Reproductive Endocrinology and Infertility Medicine Department, King Fahad Medical City, Riyadh, Saudi Arabia, Kingdom of Saudi Arabia
Received: February 25, 2015; Accepted: March 20, 2014; Published: March 31, 2014
Abstract
The presence of any three of the vertebral defects, anal atresia, cardiac defects, trachea-esophageal fistula, renal or limb anomalies describes VACTERL/VATER syndrome. In addition to these defects, patients may have other anomalies and its incidence ranges between 1 in 10,000 to 1 in 40,000 live born infants.
This is a case report for a 13-year old nulligravida who presented with primary amenorrhea and pelvic pain. Clinical examination revealed immature secondary sexual characteristics, a blind vagina and multiple skeletal defects. Ultrasound reported Hematometra, hematocolpos and right hematosalpinx. The solid structure in the lower end of the vagina represents either a thick transverse lower vaginal septum or agenesis of the lower vagina. Pelvic magnetic resonance concurred with the ultrasound findings. A skeletal survey was performed which showed S- shaped scoliosis of the thoracolumbar spine contracted deformed pelvis with bilateral hip dislocation. Trans esophageal Echocardiography reported small ASD-II, and trivial tricuspid regurge. The karyotype was 46XX. Chromosomal microarray revealed several large regions of homozygosity with at least 6% of the genome. The patient was diagnosed as mullerian hypoplasia class II according to the standard American Fertility Society in addition to multiple skeletal deformities along with large regions of homozygosity. The reconstructive surgical approach and hysterectomy options were discussed. Hysterectomy was performed.
Keywords: Primary amenorrhea; Unicornuate uterus; Vaginal agenesis; Chromosomal microarray
Introduction
The disruption of normal anatomy of the female genital tract results in congenital malformations which are a consequence of fetal embryologic mal-development of the Mullerian or paramesonephric ducts [1].
When there is a failure of one of the mullerian duct development, the result is unicornuate uterus without a rudimentary horn and when there is failure to canalize, the result is unicornuate uterus with a rudimentary horn, without proper cavity [2]. Different studies have reported the prevalence of congenital anomalies of female genital tract, ranging between 4 and 7% [3].
The presence of any three of the vertebral defects, anal atresia, cardiac defects, trachea-esophageal fistula, renal or limb anomalies describes VACTERL/VATER syndrome. In addition to these defects, patients may have other anomalies and its incidence ranges between 1 in 10,000 to 1 in 40,000 live born infants and the diagnosis is usually made when there is no clinical or lab evidence of a specific syndromes [4].
The etiology of these syndromes remains unclear to date, however new genetic research methods offer promise. Whole genome testing technologies has been the most recent laboratory tools available for clinical use. With the introduction of Single nucleotide polymorphism (SNP) arrays; regions of loss of heterozygosity (LOH) can also be detected [5].
Most of the genes demonstrate biallelic expression but some are only expressed from one allele and is also parent specific, this phenomenon is called “Genomic Imprinting”, which describes the parent of origin preferential gene expression [6]. Uniparental Disomy (UPD) is the inheritance of both chromosome’s homologues (and even part of the chromosomes) containing the imprinted genes clusters [7].
Managing these patients need a multidisciplinary approach with a gynecologist, clinical geneticist, cytogeneticist, pediatrician and a psychologist [4]. The management is usually surgical to correct many of the defects along with long term follow up and support to the patient and the family [4].
We hereof, describe a patient who has a similar presentation to VACTERAL along with Microarray analysis which revealed the presence of excessive loss of heterozygosity, > 3Mb which is above 6% of the patient’s genome and the association will be better defined in near future.
Case Presentation
A 13 year old, Saudi, single virgin female was accompanied by her mother seeking medical advice in our emergency department for severe lower pelvic pain. The patient had been to another hospital a week earlier with the same complaint, where she was diagnosed as a case of vaginal agenesis. There, an attempt was made under general anesthesia to create a connection between the uterus and the vagina, but the procedure was aborted and the patient was asked to travel abroad or seek other professional opinion.
The patient lives with her family in the suburbs of the Kingdom of Saudi Arabia, her parents are first degree cousins, and are healthy. She has 6 normal siblings. There is no family history of primary amenorrhea, congenital defects or known chromosomal anomalies. She had her menarche at the age of 14.She had no surgical problems during her life time.
The patient was on a wheel chair. Upon examination, no dysmorphic features were seen, however there was mal-alignment of her teeth. Her breast was tanner stage 2, Pubic hair was tanner 2. She had flexion deformities of the elbows with limited supination and also limited range of movement in her shoulders. She had short femur and fixed-flexion deformity of the hips. The mid-feet were stiff and had polydactyl with ten toes in the left foot out of which half were fused. She had a small vulva, normal urethra, and small labial folds; the vagina was blind and there was a dimple which represented the previous surgical incision site. Rectal exam revealed a palpable mass, 5 to 6 cm above the perineum, which represented the hematocolpos.
Her complete blood count, renal profile, liver function tests were all normal. Abdominal and Pelvis ultrasound reported normal right ovary and non-visualization of the left ovary, the uterine cavity was distended with blood(hematometra), 36 mm in Antero-posterior and 91 mm in cranio-caudal diameter, the vagina was distended with blood (hematocolpos)measuring 47 mm, and there appeared a solid structure in the lower part of the vagina, measures 20mm long and 29 mm wide (vaginal agenesis or septum), right tube was dilated with blood (hematosalpinx). Magnetic resonance imaging of abdomen and pelvis concurred with the ultrasound findings and further revealed severe degree of lumbosacral scoliosis with multiple vertebral body and presacral tissue masses (Figure 1). A skeletal survey was performed which showed S- shaped scoliosis of the thoracolumbar spine contracted deformed pelvis with bilateral hip dislocationthe left more than the right, hypo plastic left femur, and deformed, dislocated bilateral ankle joint and feet. Echocardiography revealed a small a trial septal defect (ASD) and trivial tricuspid regurgetation. Multidisciplinary meeting with 6 gynecologist suggested that hysterectomy in her case would be the best option, which was further discussed in detail with the family.
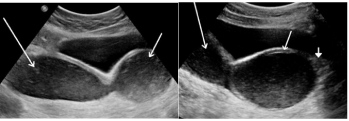
Figure 1: (a) USG pelvis image shows hematometra in a unicornuate uterus
(long arrow) and hematocolpos (short arrow).
(b) Hematomera (long arrow), Hematocolpos (short arrow) and lower 1/3rd
vaginal agenesis in the same patient (solid arrow).
Consultation with the pediatrician and the clinical geneticist indicated that this is a sporadic case, and the cytogeneticist advised parents and patient’s chromosomal and genetic testing; unfortunately the father was bedridden in another city. Chromosomes karyotype analysis of the patient was carried out on metaphase preparations following the routine cytogenetics culture and harvest methods. Twenty GTG banded metaphases on 550 band level were analyzed using the Cytovision, Applied Imaging (Lieca, Germany). Sub telomere Fluorescent in situ hybridization (FISH) was carried out to test the integrity of the telomeres ends using the Two Television (Abbott, USA) following the manufacturer’s recommendations. CMA was carried out using the CytoScan HD microarray platform (Affymetrix, USA). The resolution for this array is 30kb for deletions and 60kb for duplications. The Microarray testing was done at Mayo Medical Laboratories (Rochester, USA). Data analysis was based on the human genome build 37.1 (hg19, February 2009). Chromosomes karyotype analysis on this patient revealed 46, XX an apparently normal female karyotype. FISH analysis for the sub telomere areas was normal. Microarray analysis revealed the presence of excessive loss of heterozygosity, > 3Mb which is above 6% of the patient’s genome.
The patient was diagnosed as mullerian hypoplasia class II according to the standard American Fertility Society. Long discussion was undertaken with the mother and her brother to decide on the best possible option for the girl and they opted for hysterectomy which was performed by laparotomy with the support and consent of the family. To the best of our knowledge this is the first reported case of Unicornuate uterus with multiple skeletal deformities along with large regions of homozygosity encompassing at least 6% of the genome (Figure 2).
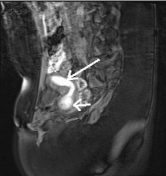
Figure 2: MR image of the same patient, verifying the mullerian anomaly,
hematometra in a unicornuate uterus (long arrow) and hematocolpos (short
arrow).
Discussion
There has been a recent report of an association between VATER/VACTERL syndrome and a unicornuate uterus and/or noncommunicating uterine horn [2]. This patient had uterine, cardiac, limb, and vertebral anomalies, in addition to dental malformation. We hereof report an additional finding of dental mal-alignment and a large area of heterozygosity of the patient’s genome after microarray testing.
This case describes an association between VATER/ VATERL syndrome and a unicornuate uterus with lower vaginal agenesis. The patient was diagnosed as mullerian hypoplasia class II according to the standard American Fertility Society [8]. Radio logical advances with multi planer cuts such as those used in MRI is necessary to diagnose such complex anomalies [9]. Diagnosing these cases need the liaison of several specialties in order to provide the patient with the best possible management options along with a long –term follow up plan. It is prudent to emphasize on the importance of multidisciplinary team approach in treating patients with multiple intricate anomalies.
It is known to us that this patient’s parents are first degree relatives however with the above laboratory work up; a possible underlying autosomal recessive disorder could not be discovered. Several chromosomes including chromosomes 6, 7, 11, 14, 15 & 20 contain areas of imprinted genes and can be sensitive to UPD [5, 7]. Table 1 is summarized version of the UPD Chromosomes associated with imprinting disorders [10].
![]()
Chromosome
Imprinted Locus
Parental Origin
Associated Disorder
Major Clinical Features
Known/Proposed Genes
6
6q24
Paternal
Diabetes Mwllitus, 6q24 related transient neonatal
IUGR, neonatal hypoglycemia
PLAG1, HYMAI
7
7p11.2-p12 and 7q32.2
Maternal
Russell-Silver
IUGR, Postnatal growth retardation, triangular facies
GRB10, MEST
11
11p15.5
Paternal (segmental)
Beckwith-Weidemann
Macrosomia, macroglossia, visceromegaly, omphalocele
IGF2, H19, CDKN1C, KCNQ1, KCNQ1OT1
Maternal
Russell-Silver
IUGR, Postnatal growth retardation, triangular facies
14
14q32.2
Maternal
Maternal UPD 14
Precocious puberty, hypotonia, joint laxity
RTL1, DLK1
Paternal
Paternal UPD 14
Skeletal abnormalities, joint contractures, intellectual disability
15
15q11-q13
Paternal
Angelman
Severe intellectual disability, speech impairment
UBE3A
Maternal
Prader-Willi
Hypotonia, hypogonadism, obesity
SNRPN, MKRN, MAGEL2, NDN, U5snoRNAs
20
20q13.3
Paternal
Pseudohypoparathyroidism type 1b
Neonatal hyperbilirobinemia, parathyroid hormone resistance
GNAS
Table 1: UPD Chromosomes Associated Imprinting Disorders.
On an array result, Certain SNP and small regions of homozygosity could be present with no clinical impact in all out bred populations, large regions of homozygosity that are restricted to a particular chromosome reflect UPD while large region of homozygosity on multiple chromosomes usually reflect parental consanguinity [8]. Long-contiguous stretches of homozygosity (LCSH) describes the areas of the genome where the uninterrupted regions of homozygosity has a copy number state of 2 and a threshold of 0.5-1Mb [10]. In general, when LCSH is observed, both the Consanguinity and the regions involved need to be compared with the imprinted gene regions (Table 1, 2). The suspicion of a recessive disorder is increased.
Consanguinity for this patient was ruled-in which explains the LCSH regions as identical by descend (IBD). Segmental LCSH has been observed in this patient (Table 2) but none of the above chromosomal regions falls among the known UPD disorders (Table 1).
![]()
Chromosome
Breakpoints
Genomic Size
1
1p13.3-p13.2
4.8
1q31.3-q32.1
5.0
2
2p24.3-p22.3
17.8
2p12-q14.2
45.3
4
4p15.1
4.0
4q27-q28.3
10.7
6
6p23-p21.31
22.2
6p16.3-q21
7.2
9
9p24.2-p23
10.0
11
11q11-q22.1
45.0
20
20q11.23-q12
4.1
Table 2: SNP array result for regions with LOH.
The complex finding of mal-aligned teeth, unicornuate uterus, absence of lower vagina, multiple skeletal defects, small ASD, may represent a new syndrome.
SNP arrays are very powerful tools in diagnostic molecular cytogenetics set-up in the detection of UPD and LOH However, areas of the genome that are not covered by the array remains questionable.
Large area of LCSH exceeding 3Mb is considered regions of IBD due consanguinity. It also raises the suspicion of an autosomal recessive disorder. SNP arrays are very powerful tools in diagnostic molecular cytogenetics set-up in the detection of UPD and LOH. However, areas of the genome that are not covered by the array remain questionable.
It has been hypothesized that for genes involved in rare recessive disorders are yet to be discovered, and that approaches of homozygosity mapping can be the approach of choice for the identification of such genes in consanguineous families. For this particular patient no clinical correlation with a specific autosomal recessive disorder was reached, which raises a question of where to go next for diagnosis of similar cases. However due to the rare nature of these syndromes a larger studies are essential [11].
References
- Grimbizis GF, Campo R. Congenital malformations of the female genital tract: the need for a new classification system. Fertil Steril. 2010; 94: 401-407.
- Nunes N, Karandikar S, Cooper S, Jaganathan R, Irani S. VATER/VACTERL syndrome (vertebra/anus/cardiac/ trachea/esophogus/radius/renal/limb anomalies) with a non-communicating functioning uterine horn and a unicornuate uterus: a case report. Fertil Steril. 2009; 91: 1957.
- Acién P, Acién M, Sánchez-Ferrer M. Complex malformations of the female genital tract. New types and revision of classification. Hum Reprod. 2004; 19: 2377-2384.
- Solomon BD. VACTERL/VATER Association. Orphanet J Rare Dis. 2011; 6: 56.
- Manning M, Hudgins L. Professional Practice and Guidelines Committee . Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010; 12: 742-745.
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001; 2: 21-32.
- Tucker T, Schlade-Bartusiak K, Eydoux P, Nelson TN, Brown L. Uniparental disomy: can SNP array data be used for diagnosis? Genet Med. 2012; .
- The American Fertility Society classifications of adnexal adhesions, distal tubal obstruction, tubal occlusions secondary to tubal ligation, tubal pregnancies, Müllerian anomalies and intrauterine adhesions. Fertil Steril. 1998; 49: 944–955.
- Troiano RN, McCarthy SM. Mullerian duct anomalies: imaging and clinical issues. Radiology. 2004; 233: 19-34.
- Kearney HM, Kearney JB, Conlin LK. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med. 2011; 31: 595-613, ix.
- Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ. Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet. 2012; 91: 275-292.