
Review Article
Sarcoma Res Int. 2014;1(1): 8.
Immunotherapy in Sarcoma: A Brief Review
Hu JS1,3*, Skeate JG2,3, Kast WM2,3 and Wong MK1,3
1Department of Medicine, Division of Medical Oncology, University of Southern California, USA
2Department of Molecular Microbiology & Immunology, University of Southern California, USA
3Norris Comprehensive Cancer Center, University of Southern California, USA
*Corresponding author: James S Hu MD, Department of Medicine, Division of Medical Oncology Norris Comprehensive Cancer Center, University of Southern California, 1441 Eastlake Ave, Los Angeles, California, USA
Received: August 26, 2014; Accepted: Sep 11, 2014; Published: Sep 18, 2014
Abstract
The systemic treatment of soft tissue sarcomas other than Gastrointestinal Stromal Tumors (GIST) has not changed for several decades. The recently demonstrated effectiveness of immune checkpoint inhibitors in melanoma has led to its application to other solid tumors with many ongoing studies that await completion. Soft tissue sarcomas possess several classes of immunogenic antigens that could provide a basis for future immunotherapy trials. The presence of Cancer Testis Antigens (CTA) and other immunogenic antigens unique to soft tissue sarcomas will be reviewed here along with a review of past studies that may shed light on the design and conduct of future immunotherapy trials in sarcoma.
Keywords: Immunotherapy; Cytokine; Osteosarcoma; Fibrosarcoma; Synovial sarcoma
Abbreviations
CTA: Cancer Testis Antigens; GIST: Gastrointestinal Stromal Tumors; STS: Soft Tissue Sarcomas; TNF: Tumor Necrosis Factor; SBRT: Stereotactic Body Radiation Therapy; HLA: Human Leukocyte Antigen; TILS: Tumor Infiltrating Lymphocytes
Introduction
Effective systemic therapy options in advanced Soft Tissue Sarcomas (STS) are limited and overall survival benefits remain elusive. Therefore, other treatment options should be explored, and one of the more promising treatment options is immunotherapy. The purpose of this review is to explore the rationale and evidence supporting the use of immunotherapy in the treatment of sarcoma.
Background
Because of the heterogeneity and relative rarity of these tumors, standard first line options using doxorubicin based chemotherapy for intermediate and high grade STS have not changed over the past two decades. Although modest responses have been documented, overall survival benefits have not improved in the advanced setting [1]. Recently, targeted therapies have proven beneficial for progression free survival after first line therapy in some sarcomas; however, this modest improvement has not translated into an overall survival benefit [2]. Thus, there is a need for improved systemic therapies in STS.
The early observations that soft tissue sarcomas can regress after an infection and, conversely, patients with immune deficiencies can develop solid tumors, including sarcomas, at a higher rate implies that the immune system can play a role in the natural history of sarcoma [3,4]. In one study, patients that have had organ allografts were more prone to developing sarcomas than the general population. In a study of 8191 organ transplant patients, 7.4% of patients who developed malignancies developed sarcoma and 1.7% developed non-Kaposi's sarcomas. Furthermore, it was observed that of the 15 sarcomas that developed in pediatric patients, five of them were of the leiomyosarcoma subtype, which is unusual in this patient population [4].
The recent FDA approval of ipilimumab in melanoma and reported efficacy in other tumor types has led to newer applications of these anti-CTLA-4 antibodies to different settings such as in the adjuvant setting or combined with radiation in the setting of metastatic disease to the brain. (NCT00636168, NCT01703507) (All national clinical trials listed in Table 1). The anti-CTLA-4 antibody Tremelimumab has shown activity in gastrointestinal malignancies and non-small cell lung cancer [5,6]. These studies and the other reported studies using anti-PD1 approaches make check point inhibitors an attractive option for other malignancies such as sarcoma.
![]()
NCT Study #
Target/Type
Treatment
Patients/Design
Endpoints
Disease
00636168
CTLA-4/antibody
Ipilimumab vs Placebo
Adjuvant
Randomized Phase III
Relapse Free Survival
Melanoma
01703507
CTLA-4/antibody
Ipilimumab + Brain Irradiation
Metastatic/Phase 1
Safety
Melanoma
00134030
Nonspecific
MAP vs MAP/IFN alpha in good response group
Adjuvant
Randomized Phase III
Event Free Survival
Osteosarcoma
00101309
Nonspecific/vaccine
Autologous tumor vaccine + EBV transformed lymphoblastoid cell line + IL-2 SC
Relapsed or refractory/
Phase 1
Safety
Ewing's Sarcoma Neuroblastoma
01968109
LAG-3 +/- PD1/ Checkpoint Inhibitor
BMS 986016 alone and with Nivolumab (BMS 936558)
Incurable/Phase 1 with expansion cohort
Safety
Solid tumors
Expansion in Melanoma/
Non small cell lung/
Gastric and Head and neck cancer
01347034
Nonspecific/ Dendritic Vaccine
External beam radiation with or without autologous dendritic cell vaccine
Neoadjuvant/Locally advanced/Phase II
Immune response
Soft Tissue Sarcoma
01241162
NY-ESO-1/MAGE-A1/MAGE-A3/Vaccine
Decitabine Priming /Dendritic cell vaccine pulsed with peptide antigen
Relapsed to prior treatment/Gross tumor not required/Age 1-17/Phase I
Tolerance to Decitabine
Osteosarcoma
Ewing's sarcoma
Rhabdomyosarcoma
Synovial sarcoma Neuroblastoma
01953900
GD2/Chimeric Antigen Receptor-T cells/Adoptive Therapy
VZV vaccine/CAR with immune stimulatory domains CD28 and OX40
GD2+ sarcoma not responsive to standard therapy
Safety
Presently only enrolling Osteosarcoma
Table 1: National Clinical Trials Cited.
Despite the lack of large clinical trial data showing definitive efficacy in sarcomas, there is data from smaller studies that show that immunotherapy has activity against certain sarcomas. In particular, adoptive immunotherapy targeted to the common testicular antigen, NY-ESO-1, on synovial sarcoma has shown activity in a small number of patients. [7]. In addition, there is a growing body of literature in sarcomas that show that PD-L1 and PD-1 expression are prevalent and prognostic in retrospective analyses [8]. These small studies along with other clinical studies from pediatric sarcomas will be reviewed here in order to form a basis for the development of immunotherapy studies.
Immunotherapy can be divided into categories based on mechanism of action. For the purposes of this review, these categories are (1) nonspecific cytokine based therapies and innate cellular stimulation, (2) immune check point inhibitors, (3) vaccine therapy (active immunization), and (4) adoptive T-cell therapy.
(1) Nonspecific Cytokine Based therapies and Innate Cellular Stimulation
This category of immunotherapy includes interferons, interleukin-2, and mifamurtide (L-MTP-PE); each of which stimulates nonspecific immune responses.
Interferons: Interferons are a group of cytokines that have a number of direct and indirect effects on tumors and the immune system. They can induce innate cellular responses by activating natural killer cells and macrophages as well as enhance expression of tumor associated antigens and MHC antigen expression on tumor cells [9,10]. In addition, interferons have been shown to have direct inhibitory effects on tumor such as apoptosis induction, growth inhibition, as well as induction of anti-angiogenesis [11]. Though activity has been demonstrated in melanoma, evidence of activity for sarcoma has been limited to osteosarcoma patients.
In two non-randomized Scandinavian studies completed between 1971 and 1990, localized osteosarcoma of mainly the extremities was treated with postoperative adjuvant interferon in two different doses. The long term survivals of these patients were between 39 to 43% which was interpreted as being promising outcomes [12]. There are few studies showing efficacy in advanced disease, although one phase II study looked at interferon alpha in advanced bone sarcoma and showed two patients having a partial response [13]. Although this data is interesting, an initial analysis of the EURAMOS adjuvant osteosarcoma study did not show a statistically significant benefit with treatment in the good prognosis group and there were a considerable number of patients that did not receive treatment [14]. This study is still ongoing with results of the other subgroups still pending. (NCT00134030).
Interleukin-2 (IL-2): Interleukin-2 was isolated based on the growth induction of activated T cells. It also induces other cytokines such as Tumor Necrosis Factor (TNF) and IL-6 which can, in part, explain the significant side effects of high dose IL-2 therapy [15]. Although unclear, the primary mechanism of action of IL-2 appears to be its induction of cytotoxic T cells and NK cells to attack tumors [16]. High dose IL-2 therapy is an FDA approved therapy in melanoma and renal cell carcinoma. In addition to its up-regulation of cytotoxic T cells, it has been shown that IL-2 can reduce T cell regulatory cells in melanoma and renal cell carcinoma patients, which may enhance the cytotoxic T cell response [17]. Clinically, these responses in renal cell carcinoma and melanoma patients have been durable despite the low rate of responses observed [18].
In sarcoma, there is little evidence of efficacy using IL-2. An analysis of 652 patients treated at the National Cancer Institute with high dose IL-2 based therapy, included six sarcoma patients, none of which responded [19]. In a more recent pediatric solid tumor study, 6 patients with advanced sarcoma were identified. There were four osteosarcoma patients treated with high dose IL-2 and two achieved a complete response lasting a median of 28 months. The two Ewing's sarcoma patients had progressive disease without evidence of activity. The study demonstrated no unusual side effects nor durable responses [20]. In soft tissue sarcoma, one report in a metastatic retroperitoneal angiosarcoma patient showed an impressive response to high dose IL-2 [21]. Therefore, high dose IL-2 may have activity in selected sarcoma subtypes.
The use of inhaled IL-2 is an attractive option since the lung is a common site of metastasis in bone and soft tissue sarcoma and may avoid the significant toxicities of systemic IL-2 therapy. In clinical studies of patients with solid tumors, inhaled IL-2 was tolerable and toxicities were minimal with responses seen in 2 of 14 renal cell carcinoma patients [22,23]. In preclinical studies of mice with metastatic osteosarcomas, inhaled IL-2 used in combination with NK cell infusion demonstrated enhanced trafficking to the lung with evidence of apoptosis of the osteosarcoma lung metastasis [24]. Furthermore, no infiltrations of other organs were observed of NK cells and no systemic toxicities were noted. It is difficult to define the future role of IL-2 therapy in sarcomas, however, there is a clinical trial ongoing using the combination of IL-2 with autologous tumor vaccine therapy in Ewing's sarcoma and Neuroblastoma. (NCT00101309)
Liposomal-Muramyl-Tripeptide Phosphotidyl-Ethanolamine (L-MTP-PE): L-MTP-PE is a liposomal derivative of the cell wall of the Bacillus Calmette Guerin vaccine (BCG). It has been shown to stimulate activation of pulmonary macrophages and circulating monocytes of the innate immune system as well as cytokines such as IL-6 and TNF [25]. In a large study of osteosarcoma patients with metastatic and localized disease, there was a statistically significant improvement in overall survival in patients who had localized disease that received L-MTP-PE compared to those who did not. In this study, 602 patients with osteosarcoma with resectable localized disease were given induction chemotherapy with methotrexate, cisplatin, and doxorubicin. Patients were randomly assigned in a two by two design to MAP with or without ifosfamide and L-MTP-PE or not to be given after surgery [26]. There were no reported imbalances between the two groups receiving or not receiving L-MTP-PE in terms of grade III or IV necrosis to induction therapy. The primary endpoint of the study was event free survival and was powered to detect a difference of 36% (HR = .64). Of the 559 patients assessable, there was a non-significant benefit in terms of EFS in the MTP-PE arm vs the non MTP-PE arm (HR= 0.8, P= .08). However, the reported overall survival was significant in favor of the group that received L-MTP-PE with a hazard ratio of .71 and an improvement in overall survival at 6 years of 78% vs 70% (P=.03). The Food and Drug Administration did not approve L-MTP-PE, in part because of concerns with comparisons of L-MTP-PE across two different chemotherapy groups. (FDA ODAC 2007) However, it has been approved in Europe and Mexico.
An analysis of the 91 osteosarcoma patients with metastatic disease in the Intergroup study 0133 mentioned above was performed looking at the 5 year event free survival and OS in patients who did or did not receive L-MTP-PE. There was a non-significant improvement in 5 year event free survival of 42 vs 26 percent with overall survival improvement of 53 vs 40 percent [27]. No significant difference could be found in median EFS and OS. Interestingly 50% of patients with resected disease were alive at two years after resection which seems to compare favorably to historical controls where patients would be expected to have an approximately 30 percent 3- year survival [28]. The role of L-MTP-PE is still controversial and future clinical trials could help clarify its role.
(2) Checkpoint Inhibition
A process wherein regulatory checkpoints are inhibited resulting in T-cell stimulation.
The recent approval of the anti-CTLA-4 antibody ipilimumab for advanced melanoma has raised interest in checkpoint inhibition as a therapy for other solid tumors. In two large randomized phase III trials, ipilimumab was found to prolong survival in patients with advanced melanoma [29,30]. Cytotoxic T-Lymphocyte-associated antigen 4 (CTLA-4) is an immunomodulatory molecule that is involved with tolerance by suppressing T cell responsiveness [31]. When CD8 cells bind to a specific antigen in an MHC restricted manner, proliferation and activation depends on co-stimulatory signals. The CD86 antigen on APC cells bind to CD28 on the T cell and stimulates up-regulation and activity, however, the T cell inhibitory molecule CTLA-4 also competes for CD86 binding. Binding of these antigens (CD86 and CTLA-4) decreases proliferation of CD4 and CD8 cells and raises the threshold for their activation. It is this CTLA-4 CD86 interaction that is disrupted by anti-CTLA-4 antibody therapy, thus allowing for unrestricted activity of CD8 effector cells [32] (Figure 1A).
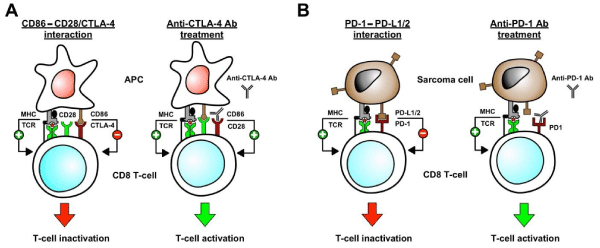
(A). CD8 Tcell activation by antigen presenting cells (APC) requires two signals. The first signal is the presentation of specific tumor associated antigenic peptides by the major histocompatibility complex class I molecules (MHC I) on APCs to the T-cell receptors (TCR) on CD8 T cells. The second signal is the interaction of CD86 molecules on the APCs with CD28 molecules on the CD8 T cells. After these two activation signals CTLA-4 is up-regulated on the CD8 T cells and starts to compete with CD28 for binding to CD86. Binding of CD86 on the APCs to CTLA-4 on the CD8 T cells leads to inactivation of CD8 T cells. An anti-CTLA-4 antibody blocks this interaction and keeps the CD8 T cells activated.
(B). Programmed death ligand 1 and 2 (PDL-1/2) molecules are expressed on the surface of sarcoma cells. Interaction between sarcoma associated PDL1/ 2 and T cell programmed death 1 (PD-1) on the CD8 T cells leads to inactivation of tumor-directed CD8 T-cells, allowing the sarcoma to evade CD8 T cell mediated killing. Use of anti-PD-1 antibodies blocks this T cell inactivation and allows the tumor-directed CD8 T-cells to remain active in destroying the sarcoma cells.
Figure 1: Anti-CTLA-4 and Anti-PD-1 antibodies are used to target T cell immune check points inhibitors in cancer immunotherapy.
(A). CD8 Tcell activation by antigen presenting cells (APC) requires two signals. The first signal is the presentation of specific tumor associated antigenic
peptides by the major histocompatibility complex class I molecules (MHC I) on APCs to the T-cell receptors (TCR) on CD8 T cells. The second signal is the
interaction of CD86 molecules on the APCs with CD28 molecules on the CD8 T cells. After these two activation signals CTLA-4 is up-regulated on the CD8 T
cells and starts to compete with CD28 for binding to CD86. Binding of CD86 on the APCs to CTLA-4 on the CD8 T cells leads to inactivation of CD8 T cells. An
anti-CTLA-4 antibody blocks this interaction and keeps the CD8 T cells activated.
(B). Programmed death ligand 1 and 2 (PDL-1/2) molecules are expressed on the surface of sarcoma cells. Interaction between sarcoma associated PDL1/
2 and T cell programmed death 1 (PD-1) on the CD8 T cells leads to inactivation of tumor-directed CD8 T-cells, allowing the sarcoma to evade CD8 T
cell mediated killing. Use of anti-PD-1 antibodies blocks this T cell inactivation and allows the tumor-directed CD8 T-cells to remain active in destroying the
sarcoma cells.
A phase II clinical trial was conducted using ipilimumab in synovial sarcoma patients whose tumors expressed the NY-ESO-1 antigen [33]. In this study, 6 patients were treated with one course of ipilimumab given at 3 mg/kg every 3 weeks for 3 doses. There were no responses according to Response Evaluation Criteria in Solid Tumors (RECIST) criteria observed and only one patient demonstrated an immune response. This study was terminated early for slow accrual, lack of activity, and lack of immune responses. It is noted that the doses in two positive melanoma trials were at 10 mg/kg for four doses instead of 3mg/kg for 3 doses as used in this synovial sarcoma trial. In addition standard RECIST criteria were used in the assessment of patients in the sarcoma trial instead of immune related response criteria (irRC). Though not reported in this study, pseudo-progression characterized by increases in measurable tumor and new lesions can complicate the interpretation of progressive disease in patients treated with immunotherapy. Therefore, part of the lack of response in this trial could be attributable to the unique pattern of responses seen in tumors subjected to immunotherapy which could be missed by standard RECIST criteria. In previous immunotherapy trials of melanoma patients, it was noted that many tumors may increase in size or develop new lesions prior to responding to treatment. By using immune related response criteria which allows for a 25% increase in tumor volume and new lesions, more accurate clinical assessment of immunotherapy effects could result [34].
The Programmed Death one (PD-1) and programmed death ligand one pathway (PD-L1) is another checkpoint pathway that can be exploited using anti-PD-1 approaches (Figure 1B). Early studies of anti PD-1 agents (MDX 1106) resulted in 3 of 39 responses. These responses in colon cancer, renal cancer, and melanoma led to a large study in melanoma [35]. The role of the PD-1/PD-L1 axis in STS and melanoma is presently being elucidated. In one retrospective study of tumor specimens from 105 cases, it was determined that the degree of PD-1 positivity in tumor infiltrating lymphocytes and PD-L1 overexpression in tumor specimens correlated with a poorer prognosis and more aggressive disease [8]. These studies suggest that PD-1 and PD-L1 staining may be viable biomarkers for prognosis. As a predictor for response using anti-PD-1 treatment strategies, PD-1 and PD-L1 staining may not be as predictive. In preclinical trials using implanted fibrosarcoma mouse models, activity of anti-PD-1 therapy was independent of PD-L1 staining [36]. Therefore, just as in melanoma, the role of PD-L1 staining as a biomarker is still largely unknown.
Other studies in fibrosarcoma rodent xenografts have shown modest activity using anti-PD-1 therapy alone, but significantly enhanced activity when combined with a dual checkpoint antibody directed at LAG-3 [37]. Therefore, although anti-PD1 therapy has not been tested in sarcoma patients, there is ample rationale for using anti-PD-1 treatment alone and possibly in combination with other checkpoint inhibitors in patients with advanced STS. There is an ongoing phase I clinical trial in solid tumors that is testing anti-LAG -3 antibody alone and combined with anti-PD-1 therapy. (NCT01968109)
(3) Vaccine (Active Immunization) Therapy, Dendritic Cell Approaches, and Generation of "Autovaccines" via the Abscopal Effect
The manipulation of tumor antigens to stimulate both humoral and cellular immune responses.
The sine qua non of an immune response is its specificity for the unique molecular target, also known as an epitope. Therefore, the search for unique molecules that would be recognized as "foreign" by the immune system is the cornerstone of development of specific immunotherapy. The identification of immune stimulating tumor antigens is therefore critically important in the development of tumor vaccines.
In studies examining the specificity of Tumor Infiltrating Lymphocytes (TILS) for melanoma antigens, surface receptors with specificity to over 50 different antigens have been identified [38]. These included both melanoma antigens as well as a wide variety of cancer testis antigens. The importance of this knowledge is that these antigens may serve as targets responsible for the specific recognition by cytotoxic T cells. The lists of such antigens in sarcomas include not only melanoma differentiation antigens and cancer testis antigens, but also mutated or other overexpressed antigens such as synovial sarcoma transcript proteins and gangliosides [39,40]. Since up to 30 percent of sarcomas may express unique fusion proteins, a variety of antigens can be exploited as targets for immunotherapy. The NY-ESO-1 antigen is a known cancer testis antigen recognized by cytotoxic T cells and is expressed in over 80% of synovial sarcomas, 100 percent of myxoid round cell liposarcomas, and in a high percentage of osteosarcomas and uterine leiomyosarcomas [41- 43]. MAGE-A3 is another cancer testis antigen that was originally discovered in melanoma and, interestingly, is expressed in some nonuterine leiomyosarcomas as well as uterine leiomyosarcomas [44].
Although molecular characterization of tumor antigens has been successful, there has been little evidence that active vaccination alone can lead to tumor regression. It appears that it may be necessary, but insufficient by itself. Use of tumor derived peptides, proteins, whole tumor cells, recombinant viruses, dendritic cells, and heat shock proteins have yielded few responses in any solid tumors. A review of over 1000 published vaccine treatment articles, showed an abysmal overall objective response rate of 3.3% [45]. Analyses of surrogate markers of benefit, such as the presence of circulating T cells and tumor infiltration by T cells, have taught us that detection of an immune response to these epitopes is clearly not linked to clinically meaningful responses. In sarcoma, one study used peptides from the unique and restricted SYT: SSX fusion gene, found in most synovial sarcomas, and demonstrated induction of peptide specific cytotoxic T cells in 4 patients, but only modest clinical benefit in one patient [40].
Resetting the balance towards immune stimulation underlies a series of studies in which vaccines were combined with a second immune stimulation signal. Augmenting the immune response after vaccination using a cytokine was attempted in a 21 patient pilot study with synovial sarcoma patients in whom an SYT-SSX fusion region peptide vaccine was given in conjunction with interferon-alpha [46]. These patients were injected subcutaneously with the vaccine and assessed for response. Although only one of 21 patients had a minor response, 6 of the patients demonstrated stable disease at 12 weeks. The duration of stable disease was not reported, so no conclusion about durability could be ascertained, however, the treatment was feasible and tolerable. The efficacy of GM-CSF based approaches in melanoma patients has been inconsistent when used as the sole therapy or combined with vaccines [47]. In one study using whole cell vaccine combined with genetically engineered tumor cells for GM-CSF expression, melanoma patients showed a significantly lower number of circulating CD8 positive cells and inferior rates of progression free survivals [48]. A phase 1 study in melanoma and sarcoma patients treated with autologous tumor vaccine that was genetically engineered to express GM-CSF showed only one patient with immunologic response to tumor vaccine and no responses in a group of 6 sarcoma patients [49]. Therefore, the effectiveness of cytokine enhanced vaccine approaches is inconclusive and future studies should include additional modifying techniques to alter the balance in favor of immune stimulation.
The Major Histocompatibility Complex (MHC), in humans called the Human Leukocyte Antigen (HLA) system is known to play a major role in tumor immunogenicity. By developing a vaccine that presents tumor antigens in an MHC restricted manner, better immune responses may be elicited. In one sarcoma study, multiple tumorassociated antigens associated with four HLA matched peptides were used to vaccinate 20 sarcoma patients in a personalized peptide vaccination approach. This personalized vaccine approach resulted in stable disease in 6 of 20 patients with the median stabilization period being 9.5 months and the longest duration of response being 35 months. This compared favorably with other studies where patients typically survived a median of 8 months and PFS was 23% at 6 months [50]. Patients that did not respond had elevated IL-6 levels during therapy. Interleukin-6 is reported to increase the concentration of Myeloid Derived Suppressor Cells (MDSC) and other immune suppressing cell subsets. Cytotoxic T-cells derived from peripheral blood mononuclear cells were noted to be enhanced in a high proportion of the patient cohort, as only 3 of 20 demonstrated peptide specific T cell responses pre-vaccination whereas 13 of 17 patients developed cytotoxic T cells post vaccination. Although it is difficult to draw conclusions from this study, it does re-emphasize the importance of MHC contributions to tumor recognition.
Other attempts at enhancing vaccine potency have centered on dendritic cell vaccines. By isolating dendritic cells and pulsing them with tumor antigens ex-vivo, enhanced immunogenic responses may be realized. The most recent example of this was performed in metastatic prostate cancer where dendritic cells were apheresed from patients and pulsed ex vivo with prostate acid phosphatase antigen linked to GM-CSF. This large study, which led to the FDA approval of Sipuleucel-T, demonstrated a low percentage of objective PSA and RECIST responses of 2.7% with no difference in PFS, however, the vaccine group had a median survival of 25.8 months vs 21.7 months for the placebo group. (p=.03) [51].
In sarcoma, dendritic cell vaccines have been used in small pilot studies. In a report from a study of solid tumors in children using pulsed dendritic cells with tumor cell lysates and Keyhole Limpet Hemocyanin (KLH), a dramatic tumor response was seen in a fibrosarcoma patient who had metastatic disease to his back and lungs. This lysate-pulsed dendritic vaccine showed no observable toxicity and an objective response in both tumor and bone [52]. Another application of dendritic cell vaccines is its use in vivo by direct injection of tumors after radiation therapy. Since local tumor irradiation had been shown to elicit immune responses when combined with direct tumor dendritic cell injection in preclinical models, a therapeutic strategy using standard fractionated radiation combined with intra-tumoral injection was completed. Finkelstein reported on the injection of dendritic cells in sarcoma patients that received radiation to their primary tumor. Immune responses were seen in 9 of 17 injected patients, and 12 of 17 remained progression free at one year [53]. It was not reported whether immune response correlated with progression free survival, however, a randomized study looking at locally advanced sarcoma patients with greater than 5 centimeter tumors is underway comparing intra-tumoral dendritic cell vaccine to no vaccine. (NCT01347034)
One of the theoretical issues with the use of the aforementioned approaches is that one is never sure that the correct epitope is being targeted. There are two approaches to this issue, one is to induce the up-regulation of tumor antigens and the other is to cause a release of these via cytolytic mechanisms such as tumor irradiation.
The use of epigenetic modifying compounds such as decitabine is an example of the first strategy. Preclinical studies show that cancer testis antigens; NY-ESO-1, LAGE-1, SSX, and MAGE-A10, can be up-regulated in sarcoma cell lines using decitabine. These in vitro cell line studies likewise show that in osteosarcoma, Ewing's sarcoma, and rhabdomyosarcoma; MAGE-A1, MAGE-A3, and NY-ESO-1 can increase up to 2500 fold under the influence of decitabine [54]. Concomitant up-regulation of MHC molecules and antigen specific CTL recognition were also demonstrated. These findings raise the possibility of enhancing an antitumor effect by priming tumors to overexpress cancer antigens using systemic agents, in essence, increasing the target volume in the presence of the targeting cells. An ongoing Phase 1 study using decitabine followed by a dendritic cell vaccine pulsed with NY-ESO-1, MAGE-A1, and MAGE-A3 in young patients with neuroblastoma, synovial sarcoma, osteosarcoma, rhabdomyosarcoma, and Ewing's sarcoma is ongoing [55]. (NCT01241162)
The work by the Oregon group led by Drs. Urba and Curti is an example of using radiation to prime the immune system such that the application of a subsequent immune stimulus, in this case high dose IL-2, results in a significant percentage of patients obtaining a meaningful response. In their study, treatment naive patients with metastatic melanoma or renal cell carcinoma underwent Stereotactic Body Radiation Therapy (SBRT) followed by high dose IL-2 [56]. Eight of 12 patients (66.6%) achieved a complete (CR) or Partial Response (PR) in a setting where mature historical data showed response rates in the 10 to 20 percent range.
This is perhaps the most vivid example of an effect that is being observed post radiation, where untreated tumor deposits distant from an irradiated tumor can show tumor response. The so-called abscopal effect exists in the realm of case reports and cohort studies. The Oregon investigators hypothesized that radiation increases tumor antigen release and changes in the tumor microenvironment such that the immune effects of IL-2 are significantly more effective in melanoma and perhaps renal cell carcinoma. Whether these effects can be seen in sarcoma is not known.
(4) Adoptive Cell Therapy
Infusion of manipulated T-cells that have been activated and/or expanded ex-vivo.
Adoptive cellular immune therapy is an immunotherapy approach that infuses immune-manipulated lymphocytes into a cancer patient in order to elicit an antitumor response. One advantage of this strategy is that the ex vivo expansion of these manipulated T cells allows for the reintroduction of large numbers of these activated cells. In addition, the absence of physiologic counterbalancing inhibitory mechanisms which naturally occur in vivo can be avoided or manipulated to favor the immune balance in favor of these infused activated T-Cells. Oftentimes, this strategy is combined with host conditioning, such as non-myeloablative chemotherapy or body radiation, which further enhances the possibility of the desired immune response [57].
This strategy is best worked out in melanoma patients. In a study of 13 patients treated with a conditioning regimen of non-myeloablative therapy and IL-2 combined with adoptive autologous T cells directed at MART antigens, 6 of 13 patients had a partial response [58]. T-cell expression of peripheral lymphocytes showed evidence of viability of infused T-cells and evidence of in vivo expansion of these cells. Biopsy of tumor samples that showed increased tumor infiltration by CD8 positive lymphocytes was evidence of T cell receptor activity towards the MART antigen.
The demonstration of effective adoptive therapy in sarcoma patients was carried out in a study using autologous T cells engineered with T-cell receptor directed at NY-ESO-1 antigen in synovial sarcoma patients. Eighty percent of synovial cell sarcomas express this antigen, and its expression was an inclusion criterion for this study. Patients were initially lympho-depleted using cyclophosphamide and fludarabine; autologous T cells genetically engineered to recognize NY-ESO-1 and HLA-A*0201 antigens were then infused. Partial responses occurred in 4 of 6 sarcoma patients, with one response durable to 18 months. This therapy was also well tolerated with no off target effects [7].
Use of isolated human T cells with receptor specificity for unique molecular signature breakpoints has not been used in studies in synovial sarcoma patients. However, the feasibility of this adoptive approach directed at the unique synovial sarcoma x break point 2 (SSX2) in melanoma cells has been tested. These clonal T cells were observed to release gamma interferon and included cell lysis in an HLA restricted manner after culture of TCR engineered Peripheral Blood Lymphocytes (PBL) with relevant tumor cell lines [59]. Although these cells were originally derived from melanoma cells of two melanoma patients, application of this technology to synovial sarcoma patients who preferentially express the SSX2 antigen is an exciting possibility.
Other sarcoma translocation breakpoint studies using adoptive approaches directed at pediatric sarcomas have been completed. Dendritic cell vaccines pulsed with specific pediatric translocation breakpoint peptides along with a peptide known as E7 (which binds to HLA-A2) were used as priming antigens and combined with infusion of autologous T-cells in a study of Ewing's patients harboring the 11:22 translocation and alveolar rhabdomyosarcoma patients harboring the 2:13 translocation. There were 52 patients entered into this study with 30 patients getting immunotherapy as consolidation treatment after maximal cytoreduction of tumor by chemotherapy, radiation therapy, or surgery. These alveolar rhabdomyosarcoma and Ewing's sarcoma patients had a 43% 5 year overall survival despite recurrent or metastatic disease. Although these results could be related to selection bias, patients generally tolerated the therapy well. Immune responses as measured by serum gamma interferon production were inconsistent and not sustained by dendritic cell vaccination. This study serves as a sound framework to study future consolidative immune therapy approaches in the minimal disease setting [60]. With future improvements in antigen immunogenicity and dendritic cell maturation processes, better immunologic effects may be seen and applied to the low disease burden setting using combined approaches of enhanced target exposure combined with adoptive T-cell therapy.
Unfortunately the HLA restriction of the above adoptive T-cell therapies limits the proportion of patients that can be effectively treated. Furthermore, tumor cells can often times lose MHC antigens thereby mitigating recognition by cytotoxic T-cells. Engineering an adoptive T-cell therapy whose response is not HLA restricted could potentially include a larger proportion of patients and recognize tumor cells that may have lost MHC antigen. Chimeric Antigen Receptor (CAR) therapy consists of the introduction of an engineered T-cell receptor with specificity to a desired antigen without the need for MHC restriction. The prototype of this strategy was attempted in neuroblastoma patients [61]. The ganglioside GD-2 antigen is a highly expressed antigen on neuroectodermal derived tumors and sarcomas. It is often expressed in various sarcomas including: osteosarcoma, rhabdomyosarcoma, Ewing's sarcoma, liposarcoma, fibrosarcoma, and leiomyosarcoma [39,62]. In a preliminary report of a phase 1 study in neuroblastoma patients, persistence of the CAR modified T cells were evident and a tumor response was seen in half the patients. T Cell-CAR therapy directed at GD-2 has also been reported in a preclinical model of Ewing's Sarcoma [63]. In this mouse study, there was a noticeable delay in tumor growth in lungs compared to control mice. The efficacy of GD-2-directed therapy in this study, though modest, may be more applicable in the setting of a lower tumor burden such as in patients that had pulmonary metastatectomy. For bulky disease the authors concluded that combined approaches aimed at tumor cytoreduction before use of CAR therapy may be the most effective application of this therapy. There have been various molecular iterations of this strategy in the development of second generation CAR engineered T-cells. The latest development has been manipulation of the co-stimulatory domains of the CAR in effort to enhance in vivo T cell expansion and activity. An anti-GD-2 study using CAR engineered T cells is ongoing using a next generation CAR T cell approach aimed at GD-2 in combination with a vaccine for sarcomas. (NCT01953900).
Conclusion
The use of cytotoxic agents remains the dominant paradigm for the treatment of advanced or metastatic bone and soft tissue sarcomas. Aside from gastrointestinal stromal tumors, the recent introduction of targeted therapy to sarcomas has only provided marginal benefits. Unfortunately, only the occasional patient with advanced stage disease can be cured with present strategies. With melanoma and renal cell carcinoma leading the way, immunotherapy has consistently demonstrated the ability to give rise to durable longterm responses, some of which are complete, in a subset of patients. Because sarcomas possess unique genetic changes and immunogenic antigens, immunotherapeutic strategies may be particularly fruitful in these conditions. In addition, the biological heterogeneity of sarcomas will not likely yield a unified molecular pathway that could be exploited to treat a large percentage of sarcomas, thus emphasizing the need for a therapy that may be applicable to a higher proportion of sarcoma patients. At this time, the unique confluence of both checkpoint inhibitors along with the prevalence of cancer testis antigen expression in many adult sarcomas provides a gateway to test immunotherapy strategies in sarcoma. Other strategies that include cytokines, other checkpoint inhibitors, vaccination, and adoptive T cell transfer- all of which can be given singly or in combination are currently under study. One of the major impediments has been the plethora of small, under-powered clinical trials in this area. Through collaborative efforts of the entire sarcoma community, definitive answers can be realized that may change the paradigm of treatment of a disease that has witnessed few major breakthroughs.
Acknowledgement
WM Kast holds the Walter A. Richter Cancer Research Chair. Contributions from the Karl H. and Ruth M. Balz Trust are also gratefully acknowledged. This review has been made possible by support of the Norris Comprehensive Cancer Center NCI support grant 5 P30 CA 014089-39. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
References
- Italiano A, Mathoulin-Pelissier S, Cesne AL, Terrier P, Bonvalot S, Collin F, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011; 117: 1049-1054.
- van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012; 379: 1879-1886.
- Lindberg RD, Martin RG, Romsdahl MM, Barkley HT Jr. Conservative surgery and postoperative radiotherapy in 300 adults with soft-tissue sarcomas. Cancer. 1981; 47: 2391-2397.
- Penn I. Kaposi's sarcoma in transplant recipients. Transplantation. 1997; 64: 669-673.
- Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2010; 28: 3485-3490.
- Zatloukal P, Heo DS, Park K, Kang J, Butts C, Bradford D, et al. Randomized phase II clinical trial comparing tremelimumab (CP-675,206) with best supportive care (BSC) following first-line platinum-based therapy in patients (pts) with advanced non-small cell lung cancer (NSCLC). In: American Society of Oncology Proceedings. Chicago Illinois. 2009; 27: 15s.
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011; 29: 917-924.
- Kim JR, Moon YJ, Kwon KS, Bae JS, Wagle S, Kim KM, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS One. 2013; 8: e82870.
- Brassard DL, Grace MJ, Bordens RW. Interferon-alpha as an immunotherapeutic protein. J Leukoc Biol. 2002; 71: 565-581.
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997; 15: 749-795.
- Whelan J, Patterson D, Perisoglou M, Bielack S, Marina N, Smeland S, et al. The role of interferons in the treatment of osteosarcoma. Pediatr Blood Cancer. 2010; 54: 350-354.
- Müller CR, Smeland S, Bauer HC, Saeter G, Strander H. Interferon-alpha as the only adjuvant treatment in high-grade osteosarcoma: long term results of the Karolinska Hospital series. Acta Oncol. 2005; 44: 475-480.
- Edmonson JH, Long HJ, Frytak S, Smithson WA, Itri LM. Phase II study of recombinant alfa-2a interferon in patients with advanced bone sarcomas. Cancer Treat Rep. 1987; 71: 747-748.
- Bielack SS, Whelan J, Smeland S, Marina N, Hook J, Jovic G, et al. MAP plus maintenance pegylated interferon a-2b (MAPIfn) versus MAP alone in patients with resectable high-grade osteosarcoma and good histologic response to preoperative MAP: First results of the EURAMOS-1 "good response" randomization. Abstract no. LBA10504 in Proceedings of American Society of Clinical Oncology. Chicago, Illinois. 2013; 31.
- Gemlo BT, Palladino MA Jr, Jaffe HS, Espevik TP, Rayner AA. Circulating cytokines in patients with metastatic cancer treated with recombinant interleukin 2 and lymphokine-activated killer cells. Cancer Res. 1988; 48: 5864-5867.
- Lotze MT, Grimm EA, Mazumder A, Strausser JL, Rosenberg SA. Lysis of fresh and cultured autologous tumor by human lymphocytes cultured in T-cell growth factor. Cancer Res. 1981; 41: 4420-4425.
- Cesana GC, DeRaffele G, Cohen S, Moroziewicz D, Mitcham J, Stoutenburg J, et al. Characterization of CD4+CD25+ regulatory T cells in patients treated with high-dose interleukin-2 for metastatic melanoma or renal cell carcinoma. J Clin Oncol. 2006; 24: 1169-1177.
- Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994; 271: 907-913.
- Rosenberg SA, Lotze MT, Yang JC, Aebersold PM, Linehan WM, Seipp CA, et al. Experience with the use of high-dose interleukin-2 in the treatment of 652 cancer patients. Ann Surg. 1989; 210: 474-484.
- Schwinger W, Klass V, Benesch M, Lackner H, Dornbusch HJ, Sovinz P, et al. Feasibility of high-dose interleukin-2 in heavily pretreated pediatric cancer patients. Ann Oncol. 2005; 16: 1199-1206.
- Noguchi G, Ota J, Ishigaki H, Onuki T, Kato Y, Moriyama M. [A case of retroperitoneal angiosarcoma effectively treated with recombinant interleukin-2]. Nihon Hinyokika Gakkai Zasshi. 2012; 103: 697-703.
- Heinzer H, Mir TS, Huland E, Huland H. Subjective and objective prospective, long-term analysis of quality of life during inhaled interleukin-2 immunotherapy. J Clin Oncol. 1999; 17: 3612-3620.
- Lorenz J, Wilhelm K, Kessler M, Peschel C, Schwulera U, Lissner R, et al. Phase I trial of inhaled natural interleukin 2 for treatment of pulmonary malignancy: toxicity, pharmacokinetics, and biological effects. Clin Cancer Res. 1996; 2: 1115-1122.
- Guma SR, Lee DA, Yu L, Gordon N, Hughes D, Stewart J, et al. Natural killer cell therapy and aerosol interleukin-2 for the treatment of osteosarcoma lung metastasis. Pediatr Blood Cancer. 2014; 61: 618-626.
- Kleinerman ES, Jia SF, Griffin J, Seibel NL, Benjamin RS, Jaffe N. Phase II study of liposomal muramyl tripeptide in osteosarcoma: the cytokine cascade and monocyte activation following administration. J Clin Oncol. 1992; 10: 1310-1316.
- Meyers PA, Schwartz CL, Krailo MD, Healey JH, Bernstein ML, Betcher D, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children's Oncology Group. J Clin Oncol. 2008; 26: 633-638.
- Chou AJ, Kleinerman ES, Krailo MD, Chen Z, Betcher DL, Healey JH, et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children's Oncology Group. Cancer. 2009; 115: 5339-5348.
- Aljubran AH, Griffin A, Pintilie M, Blackstein M. Osteosarcoma in adolescents and adults: survival analysis with and without lung metastases. Ann Oncol. 2009; 20: 1136-1141.
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363: 711-723.
- Robert C, Thomas L, Bondarenko I, O'Day S, M D JW, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011; 364: 2517-2526.
- Sansom DM, Walker LS. The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T-cell biology. Immunol Rev. 2006; 212: 131-148.
- Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006; 18: 206-213.
- Maki RG, Jungbluth AA, Gnjatic S, Schwartz GK, D'Adamo DR, Keohan ML, et al. A Pilot Study of Anti-CTLA4 Antibody Ipilimumab in Patients with Synovial Sarcoma. Sarcoma. 2013; 2013: 168145.
- Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009; 15: 7412-7420.
- Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010; 28: 3167-3175.
- Wei S, Shreiner AB, Takeshita N, Chen L, Zou W, Chang AE. Tumor-induced immune suppression of in vivo effector T-cell priming is mediated by the B7-H1/PD-1 axis and transforming growth factor beta. Cancer Res. 2008; 68: 5432-5438.
- Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012; 72: 917-927.
- Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001; 411: 380-384.
- Chang HR, Cordon-Cardo C, Houghton AN, Cheung NK, Brennan MF. Expression of disialogangliosides GD2 and GD3 on human soft tissue sarcomas. Cancer. 1992; 70: 633-638.
- Kawaguchi S, Wada T, Ida K, Sato Y, Nagoya S, Tsukahara T, et al. Phase I vaccination trial of SYT-SSX junction peptide in patients with disseminated synovial sarcoma. J Transl Med. 2005; 3: 1.
- Jungbluth AA, Antonescu CR, Busam KJ, Iversen K, Kolb D, Coplan K, et al. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int J Cancer. 2001; 94: 252-256.
- Pollack SM, Jungbluth AA, Hoch BL, Farrar EA, Bleakley M, Schneider DJ, et al. NY-ESO-1 is a ubiquitous immunotherapeutic target antigen for patients with myxoid/round cell liposarcoma. Cancer. 2012; 118: 4564-4570.
- Jungbluth AA, Chen YT, Stockert E, Busam KJ, Kolb D, Iversen K, et al. Immunohistochemical analysis of NY-ESO-1 antigen expression in normal and malignant human tissues. Int J Cancer. 2001; 92: 856-860.
- Ayyoub M, Taub RN, Keohan ML, Hesdorffer M, Metthez G, Memeo L, et al. The frequent expression of cancer/testis antigens provides opportunities for immunotherapeutic targeting of sarcoma. Cancer Immun. 2004; 4: 7.
- Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004; 10: 909-915.
- Kawaguchi S, Tsukahara T, Ida K, Kimura S, Murase M, Kano M, et al. SYT-SSX breakpoint peptide vaccines in patients with synovial sarcoma: a study from the Japanese Musculoskeletal Oncology Group. Cancer Sci. 2012; 103: 1625-1630.
- Kaufman HL, Ruby CE, Hughes T, Slingluff CL Jr. Current status of Granulocyte-Monocyte- Colony Stimulating Factor in the Immunotherapy of Melanoma. J Immunother Cancer. 2014; 2: 20.
- Slingluff CL Jr, Petroni GR, Olson WC, Smolkin ME, Ross MI, Haas NB, et al. Effect of granulocyte/macrophage colony-stimulating factor on circulating CD8+ and CD4+ T-cell responses to a multipeptide melanoma vaccine: outcome of a multicenter randomized trial. Clin Cancer Res. 2009; 15: 7036-7044.
- Mahvi DM, Shi FS, Yang NS, Weber S, Hank J, Albertini M, et al. Immunization by particle-mediated transfer of the granulocyte-macrophage colony-stimulating factor gene into autologous tumor cells in melanoma or sarcoma patients: report of a phase I/IB study. Hum Gene Ther. 2002; 13: 1711-1721.
- Takahashi R, Ishibashi Y, Hiraoka K, Matsueda S, Kawano K, Kawahara A, et al. Phase II study of personalized peptide vaccination for refractory bone and soft tissue sarcoma patients. Cancer Sci. 2013; 104: 1285-1294.
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010; 363: 411-422.
- Geiger J, Hutchinson R, Hohenkirk L, McKenna E, Chang A, Mulé J. Treatment of solid tumours in xchildren with tumour-lysate-pulsed dendritic cells. Lancet. 2000; 356: 1163-1165.
- Finkelstein SE, Iclozan C, Bui MM, Cotter MJ, Ramakrishnan R, Ahmed J, et al. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo-adjuvant treatment of high-risk soft tissue sarcoma patients. Int J Radiat Oncol Biol Phys. 2012; 82: 924-932.
- Krishnadas DK, Bao L, Bai F, Chencheri SC, Lucas K. Decitabine facilitates immune recognition of sarcoma cells by upregulating CT antigens, MHC molecules, and ICAM-1. Tumour Biol. 2014; 35: 5753-5762.
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008; 14: 1264-1270.
- Seung SK, Curti BD, Crittenden M, Walker E, Coffey T, Siebert JC, et al. Phase 1 study of stereotactic body radiotherapy and interleukin-2--tumor and immunological responses. Sci Transl Med. 2012; 4: 137ra74.
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008; 8: 299-308.
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002; 298: 850-854.
- Abate-Daga D, Speiser DE, Chinnasamy N, Zheng Z, Xu H, Feldman SA, et al. Development of a T cell receptor targeting an HLA-A*0201 restricted epitope from the cancer-testis antigen SSX2 for adoptive immunotherapy of cancer. PLoS One. 2014; 9: e93321.
- Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res. 2008; 14: 4850-4858.
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011; 118: 6050-6056.
- Ahmed M, Cheung NK. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett. 2014; 588: 288-297.
- Liebsch L, Kailayangiri S, Beck L, Altvater B, Koch R, Dierkes C, et al. Ewing sarcoma dissemination and response to T-cell therapy in mice assessed by whole-body magnetic resonance imaging. Br J Cancer. 2013; 109: 658-666.