
Research Article
J Schizophr Res. 2024; 11(1): 1048.
Levels of AKTIP and TUBA1B mRNA in the Frontal Pole from People with Schizophrenia: Pathophysiological and Methodological Considerations
Snelleksz M1,2*; Dean B1,2
¹The Molecular Psychiatry Laboratory, The Florey Institute of Neuroscience and Mental Health, Parkville, Victoria, Australia
²The Florey Department of Neuroscience and Mental Health, The University of Melbourne, Victoria, Australia
*Corresponding author: Snelleksz M The Florey Institute of Neuroscience and Mental Health, The University Melbourne, 30 Royal Parade, Parkville, Victoria 3052, Australia. Email: megan.snelleksz@florey.edu.au
Received: February 01, 2024 Accepted: March 12, 2024 Published: March 19, 2024
Abstract
qPCR has been used to validate results from studies of the transcriptome in postmortem brain and blood from people with schizophrenia. qPCR has specific methodological limitations which we now consider in our ongoing study of two genes that we have found altered in the frontal pole (BA 10) from people with schizophrenia using expression microarrays.
qPCR was used to measure levels of RNA of 3 possible reference genes (TFB1M, GAPDH and SKP1) and to validate changed levels in 2 genes of interest (AKTIP and TUBA1B) which were identified in an expression microarray, in BA 10 from people with schizophrenia. Our study was made more novel because we were able to divide the subjects in our schizophrenia cohort into those who were, or were not (non-MRDS), in a subtype defined by these individuals having a marked loss of cortical muscarinic M1 receptors (Muscarinic Receptor Deficit Schizophrenia (MRDS)).
Compared to controls, there were no significant differences in levels of RNA for AKTIP and TUBA1B in BA 10 from people with schizophrenia, MRDS and non-MRDS.
Our current qPCR findings contrast our previous findings using expression microarrays which showed lower levels of RNA for AKTIP and TUBA1B in BA 10 from people with schizophrenia. It is also notable that we found lower levels of TUBA1B protein in BA 10 from people with schizophrenia and higher levels of AKTIP protein in people with schizophrenia due to higher levels of that protein in MRDS. We were unable to identify any methodological issues to account for differences in our qPCR and expression microarray data. We therefore conclude that caution is needed in assessing RNA data from different methodologies when studying the molecular pathology of schizophrenia and it cannot be assumed that changes in RNA will mirror changes in protein translated from that RNA in the human brain.
Keywords: Schizophrenia; Frontal pole; qPCR; Transcriptome; Subtypes
Introduction
Schizophrenia is a serious psychiatric disorder with a complex molecular pathology that affects DNA transcription into coding [1] and non-coding [2] RNA. Changes in levels of coding and non-coding RNA in brain and blood have been reported using high-throughput transcriptomic technologies such as gene expression arrays [3] and RNA Sequencing (RNA-seq) [4]. In many of these transcriptomic studies, Quantitative Polymerase Chain Reaction (qPCR) has been used to validate the transcriptomic data as it is quantitative, reproducible and relatively rapid [5].
There are a number of issues that must be considered in using qPCR to measure RNA levels that include the consistency of sample storage, sample preparation, the need to normalise data to levels of reference genes, primer design and statistical analysis [6]. Another limitation is the potential to exhaust reagents when amplifying genes with low copy numbers [7]. The need to normalise data to levels of reference genes adds another level of complexity as the criteria for a reference gene are that their levels do not vary between different biological samples, are not affected by any experimental procedure or disease aetiologies [6]. Notably, we examined levels of RNA for six potential reference genes in Brodmann’s Area (BA) 8, 9 and 44 from people with schizophrenia and controls to show none of these genes fulfilled all of the requirements of a reference gene [8]. These, and other data [9], suggest that the selection of reference genes is critical in using qPCR to validate data from other technologies that measure levels of RNA.
Acknowledging the potential problems in using qPCR, we have been looking to further explore data we obtained using expression microarrays and tissue from the frontal pole (BA 10) from people with schizophrenia. These data showed that, compared to controls, there were 566 changes in levels of coding and non-coding RNA in BA 10 compared to 65 and 40 changes in BA 9 and BA 33, respectively [10]. This finding was of particular interest as there is mounting evidence of dysfunction at a molecular, cellular, structural, regional connectivity and functional level in BA 10 from people with schizophrenia [11]. To determine if the changes in levels of coding RNA in BA 10 from people with schizophrenia could have functional biochemical consequences, we have begun to measure levels of the proteins they encode because multiple checks and balances controlling gene translation means changes in levels of coding RNA do not necessarily correspond to changes in levels of their encoded proteins [12]. In the first of these studies, we showed a 61% decrease in tubulin alpha 1b (TUBA1B) mRNA equated to an 18.6% decrease in the level of protein in BA 10 from people with schizophrenia [10, 13].
It is now accepted that understanding the molecular pathology of schizophrenia will require the study of subtypes, based on biologically defined criteria, within that syndrome [14]. We are leaders in this endeavour having defined a subtype within schizophrenia which can be defined by a person with the disorder having a level of [3H]pirenzepine binding to the muscarinic M1 receptors in BA 9 = 110 fmol / mg estimated tissue equivalents [15] which we have termed the Muscarinic Receptor Deficit Subgroup (MRDS) within schizophrenia. Unfortunately, there were insufficient people with MRDS in our transcriptomic studies in BA 10 to determine if any changes in levels of coding or non-coding RNA were specific or more profound in those within the subgroup. It is therefore of interest that, compared to controls, we have found that there are higher levels (41%) of AKT-interacting protein in BA 10 from people with schizophrenia whereas levels of AKTIP RNA were lower (-23%) in those with the disorder. Significantly, the higher levels of AKTIP protein were only detectable in those with MRDS (+54%) with levels of the protein not being different from controls in those with schizophrenia who were not part of the subtype (non-MRDS) [16]. This raised the question as to whether levels of AKTIP mRNA were only higher in those with MRDS.
Our microarray study did not include sufficient cases to allow us to separate our data into MRDS and non- MRDS for analyses. Thus, acknowledging potential methodological limitations, we decided to use qPCR to measure AKTIP and TUBA1B mRNA in a larger cohort of people with schizophrenia than were used in our microarray study, 50% of whom were classified as MRDS.
Methods
Human Brain Collection and Processing
The tissue for this study was collected following approval from the Ethics Committee of the Victorian Institute of Forensic Medicine and after consent from the nearest next of kin.
Cadavers whose death was witnessed or who were known to be alive within 5 hours of being found deceased were stored at low temperatures until such time that the brain could be removed at autopsy and the left hemisphere processed according to a standardized procedure [17] and stored at -80°C. The cool temperatures are intended to minimize autolysis [18] and the pH was subsequently measured to ensure that the tissue was appropriately preserved [19].
On all cases, a DSM-IV psychiatric diagnosis was agreed upon by 2 psychiatrists and a psychologist [20], utilizing the comprehensive case history collected with the Diagnostic Instrument for Brain Studies [21]. In the same manner, cases without a psychiatric diagnosis were labelled as controls and were matched to psychiatric cases based on age (± 2.5 years) and sex. For all cases, the postmortem interval (PMI) was calculated as the time from autopsy to death, or the midway point between the time a donor was last known to be alive and was found dead. For cases with a diagnosis of schizophrenia, the duration of illness (DI) was calculated as the time from the first record of symptoms to death.
Lifetime Exposure to Antipsychotic drug dose standardized to chlorpromazine equivalents (LEAP) and the Final Recorded Antipsychotic Drug Dose standardized to chlorpromazine equivalents (FRADD) was also calculated.
RNA Extraction and DNase Treatment
RNA was extracted from 36 people with schizophrenia and 18 controls using the Qiagen RNeasy kit. By experimental design, the schizophrenia cohort (n=36) contained an equal number of MRDS (n=18) and non-MRDS (n=18).
Frozen tissue was homogenized in TRIzol reagent (1 mL/ 100 mg) using a chilled glass-Teflon homogenizer then transferred to a 1.7 mL tube to incubate for 5 min at room temperature. Chloroform (0.2 mL chloroform/ 1 mL TRIzol) was added and shaken vigorously for 15 sec then allowed to incubate for 3 min at room temperature before centrifuging at 12000 g for 15 min at 4°C. The upper, aqueous phase was transferred to a new tube and gently mixed with an equal volume of 70% ethanol. The sample, 700 μL at a time, was transferred to a RNeasy spin column in a 2 mL collection tube and centrifuged at 8000 g for 15 sec at room temperature, discarding the flow-through.
To wash the spin column membrane, 350 μL of buffer RW1 was added and centrifuged at 8000 g for 15 sec. The spin column membrane was incubated for 15 min in a pre-mixed solution of 70 μL buffer RDD and 10 μL DNase I stock. 350 μL of buffer RW1 was then added prior to centrifuging at 8000 g for 15 sec. 500 μL buffer RPE was added to the spin column and centrifuged at 8000 g for 15 sec then repeated with another 500 μL buffer RPE for a 2 min centrifugation. The flow-through was discarded following each centrifugation.
Finally, to elute the RNA, the spin column was placed into a new 1.7 mL collection tube. 35 μL RNase-free water was added directly to the spin column membrane and centrifuged at 8000 g for 1 min. The eluted RNA in the collection tube was immediately chilled on ice then stored in aliquots at -80°C. The concentration and quality of the extracted RNA was determined by the Nanodrop Spectrophotometer (Thermo Scientific).
Checking RNA samples for genomic DNA contamination
2 μg of each RNA sample was combined with 5 μL 10x RT-PCR buffer (100 mM Tris-HCl, 500 mM KCl, 15 mM MgCl2), 2.5 μL 2.5 mM dNTP mix (10 μL each of 100mM dATP, dCTP, dGTP and dTTP, up to 400 μL dH2O), 2.5 μL 5 μM primer mix (10 μL 100mM forward primer: TTCCGCAAGTTCACCTACC, 10 L 100mM reverse primer: CGGGCCGGCCATGCTTTACG, up to 200 μL dH2O), 0.25 μL Taq DNA polymerase and made up to 50 μL in DEPC-treated dH2O. A negative control tube contained the same mix in the absence of RNA and a positive control tube contained 2 μL of genomic DNA instead of RNA.
The PCR was run under the following conditions: 95°C for 1 minute, 30 cycles [94°C for 30 seconds, 58°C for 30 seconds, 72°C for 30 seconds], 72°C for 5 minutes. Following the PCR, 20 μL of each PCR sample was mixed with 4 μL gel loading dye on a 1% agarose gel alongside 5 μL of 100 Base Pair (bp) ladder and run at 100 V for approx. 1 hour. The sample was considered free of genomic DNA contamination if the 361bp ladder was present in the positive control lane but absent in the sample lanes and if the negative control lane was clear of any bands.
First Strand cDNA Synthesis
2 μg RNA was combined with 2 μL of 10x RT-PCR buffer, 4 μL of 2.5 mM dNTP mix, 1 μL of 50 M oligo(dT) primers, 1 μL of 50 μM random decamers, 1 μL of SUPERase•In™ RNase Inhibitor (20 U/μL), 1 μL of MMLV-RT (100U/μL) and made up to 20 μL in DEPC-treated dH2O. The samples were incubated at 44°C for 1 hour, 92°C for 10 minutes then put on ice for 2 minutes before briefly centrifuging and storing at -20°C.
qPCR
Bio-Rad PrimePCR primers were used for AKTIP (PrimePCR SYBR Green Assay: AKTIP, Human, Cat# qHsaCID0011161) and the potential reference genes TFB1M (PrimePCR Probe Assay: TFB1M, Human, Cat# qHsaCEP0058019), GAPDH (PrimePCR SYBR Green Assay: GAPDH, Human, Cat# qHSACEP0041396) and SKP1 (PrimePCR SYBR Green Assay: SKP1, Human, Cat# qHsaCIP0027663). A custom Invitrogen standard primer was used for TUBA1B (906F – CTTCAACACCTTCTTCAG, 980R – TGTCAGGTCAACATTCAG).
For AKTIP, TFB1M, GAPDH and SKP1, 2 μL of cDNA from each sample was mixed 1.25 μL of 20x Bio- Rad PrimePCR, 12.5 μL of Bio-Rad SsoAdvanced Universal SYBR® Green Supermix and 9.25 μL DEPC- treated dH2O. For TUBA1B, 2 μL of cDNA from each sample was mixed with 4 μL Invitrogen standard primer, 25 μL of Bio-Rad SsoAdvanced Universal SYBR® Green Supermix and 17 μL DEPC-treated dH2O. Every plate contained a quality control case, standard curve and non-template control which contained DEPC-treated dH2O instead of cDNA. All samples were run in triplicate.
The QIAgility automatic liquid handler was used to prepare the reactions in the iQ™ 96-Well PCR Plates (Bio-Rad Cat# 2239441). The plates were sealed with Microseal 'B' PCR Plate Sealing Film (Bio-Rad Cat #MSB1001) and placed in the Bio-Rad iQ5 Real-Time PCR Detection System.
For AKTIP, TFB1M, GAPDH and SKP1, the qPCR cycling conditions were: 95°C for 2 min, 40 cycles [95°C for 5 sec, 60°C for 30 sec] and 65-95°C increasing at 0.5°C increments for 5 sec/increment. For TUBA1B, the qPCR cycling conditions were: 95°C for 3 min, 95°C for 10sec, 40 cycles [57°C for 30sec, 72°C for 30 sec, 95°C for 1 min], 55°C for 1 min and 55-95°C increasing at 0.5°C increments for 10 sec/increment.
The baseline threshold was set to 100 for each run to obtain consistency between plates. It was ensured that the efficiency of each reaction was between 95-105%, R2 was >0.98, the melt curve had only a single product, the triplicates were consistent, and the non-template control was at least 5 cycles fewer than the samples.
Data Processing and Statistics
As we have done previously [8], data for all genes were first analysed at the levels of threshold cycle corrected for efficiency of amplification (relative quantities) and after being standardized across plates relative to the quality control sample (standardized quantities). Finally, levels of AKTIP and TUBA1B data were analysed after being normalized to the geometric mean of the reference genes that did not vary with diagnosis.
The D’Agostino & Pearson test [22] was used to determine that all data were normally distributed. The students t-test was used for analyses between the syndrome of schizophrenia and controls, and a one-way ANOVA was used for analyses between the schizophrenia subgroups (MRDS and non-MRDS) and controls. Differences in sex and death by suicide between groups were analysed using a Chi-square test and correlations between the demographic variables and AKTIP and TUBA1B were measured using Pearson’s correlation coefficient. All statistical analyses were performed using GraphPad Prism (GraphPad Software, Boston, Massachusetts USA).
Results
Demographic Variables
There were no significant differences in sex, age, pH, PMI or brain weight at the syndrome or subgroup level between people with schizophrenia, MRDS or non-MRDS and controls (Table 1, Supplementary Table 1). There were no significant differences in DI, FRADD, LEAP or rates of death by suicide between MRDS and non- MRDS (Table 1, Supplementary Table 1). Pearson’s correlation coefficient determined no significant correlations between AKTIP or TUBA1B and any of the demographic variables (Supplementary Table 2).
![]()
Sex
Age (yr)
pH
PMI (hr)
Brain Weight (g)
Suicide
DI (yr)
FRADD
LEAP
AKTIP
TUBA1B
Controls vs Schizophrenia
t or c2
2.36
0.64
1.19
0.29
1.3
0.76
0.61
d.f.
1
52
52
52
48
52
52
p
0.12
0.53
0.24
0.77
0.2
0.45
0.54
Controls vs MRDS vs non-MRDS
F or c2
2.51
0.22
0.91
0.40
0.88
0.67
0.47
d.f.
2
2,51
2,51
2,51
2,47
2,51
2,51
p
0.29
0.80
0.41
0.68
0.42
0.52
0.63
MRDS vs non-MRDS
t or c2
1.03
0.52
1.18
1.31
d.f.
1
34
32
34
p
0.31
0.61
0.25
0.20
DI: Duration of Illness, FRADD: Final Recorded Antipsychotic Drug Dose as chlorpromazine equivalents, LEAP: Lifetime Exposure to Antipsychotic drug dose as chlorpromazine equivalents, MRDS: Muscarinic Receptor Deficit Schizophrenia, PMI: Postmortem Interval. Schizophrenia represents MRDS and non- MRDS combined.
Table 1: Demographic, brain collection, pharmacological data, and levels of AKTIP and TUBA1B RNA, normalised to the geometric mean of two reference genes for the cases used in this study.
Reference Genes
At the syndrome level, levels of GAPDH (relative quantities: t(52)=0.27, p=0.79; transformed: t(52)=0.05, p=0.96), TFB1M (raw: t(52)=0.07, p=0.94; transformed: t(52)=0.20, p=0.84) or SKP1 (relative quantities: t(52)=0.88, p=0.38; transformed: t(52)=0.73, p=0.47) RNAs did not differ with diagnosis (Figure 1A). Dividing schizophrenia into MRDS and non-MRDS did not show any differences in levels of GAPDH (relative quantities: F(2,51)=0.87, p=0.61; transformed: F(2,51)=0.33, p=0.72), TFB1M (relative quantities: F(2,51)=1.17, p=0.32; transformed: F(2,51)=0.52, p=0.60) or SKP1 (relative quantities: F(2,51)=0.16, p=0.85; transformed: F(2,51)=0.07, p=0.93) with diagnosis (Figure 1A).
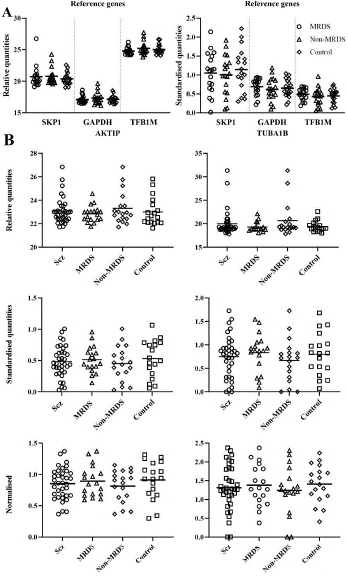
Figure 1: Levels of RNA in Brodmann’s area 10, expressed as relative quantities and standardized to controls, for S-phase Kinase associated Protein 1 (SKP1), Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) and transcription factor B1, mitochondrial (TFB1M) (A) and levels of RNA for AKT interacting protein (AKTIP) and tubulin alpha 1b (TUBA1B), expressed as relative quantities, standardised to controls and normalized to the geometric mean of two reference genes (B), from schizophrenia (Scz), muscarinic receptor deficit schizophrenia (MRDS), non-MRDS, and controls.
Although SKP1 RNA did not differ between groups, the transformed data for this RNA showed greater variability than the data from the other reference genes (GAPDH and TFB1M) (Figure 1A) and the genes of interest (AKTIP and TUBA1B) (Figure 1B). Hence, to avoid any concerns about the gene normalisation process, the geometric mean of TFB1M and GAPDH was used in the normalisation process.
AKTIP and TUBA1B
There were no significant differences between levels of AKTIP RNA in BA 10 from controls and people with schizophrenia (relative quantities: t(52)=0.19, p=0.85; transformed: t(52)=0.64, p=0.52; normalised: t(52)=0.76, p=0.45) or people with MRDS and non-MRDS (relative quantities: F(2,51)=1.20, p=0.31; transformed: F(2,51)=0.46, p=0.63; normalised: F(2,51)=0.52), p=0.67) (Figure 1B).
There were no significant differences between levels of TUBA1B RNA in BA 10 from controls and people with schizophrenia (relative quantities: t(52)=0.87, p=0.39; transformed: t(52)=0.40, p=0.69; normalised: t(52)=0.61, p=0.54) or people with MRDS and non-MRDS (relative quantities: F(2,51)=2.05, p=0.14; transformed: F(2,51)=0.73, p=0.49; normalised: F(2,51)=0.47, p=0.63) (Figure 1B, Table 1, Supplementary Table 1).
Discussion
This study shows that, compared to controls, levels of AKTIP and TUBA1B RNA do not differ in BA 10 from people with schizophrenia, MRDS or non-MRDS. This contrasts to our findings on the same cases using expression microarrays which show that levels of RNA for AKTIP (-23%) and TUBA1B (-61%) are lower in people with schizophrenia (we do not have data on MRDS and non-MRDS). In addition, at the level of protein in BA 10, we have shown that levels of AKTIP are higher in schizophrenia due to an increase in that protein in people with MRDS [16]. By contrast, TUBA1B is lower in schizophrenia whether or not a case is classified as MRDS or non-MRDS [13]. Hence, this study raises issues as to whether the technologies used to measure RNA in the human brain are giving apparent discrepant results.
The issue of non-reproducibility of findings in biological psychiatry is recognised as an impediment to increasing understanding of the pathophysiologies of these disorders [23]. It is therefore worthwhile to attempt to understand why we obtained different outcomes in measuring the level of RNA for two genes in BA 10 from people with schizophrenia. Notably, one difference between the analysis of qPCR data and high-throughput RNA data in schizophrenia is the use of reference genes to normalize qPCR data [24]. We have argued that, due to the complexity of gene expression across the human brain, no gene can fulfill all the criteria required of a reference gene [8]. However, the reference genes used for normalization for this study did not differ with diagnosis within BA 10 and had low levels of intra-personal variability making them acceptable for use in our study. Moreover, our findings comparing levels of RNA in controls to that in schizophrenia did not differ using either minimally derived or normalized data which indicates, in this study, the use of reference genes was not a significant variable.
In addition to reference gene selection, the outcomes from using qPCR to measure RNA levels can be influenced by sample preparation, sample storage, primer design and statistical analysis [6]. In our studies using expression microarrays and qPCR, RNA was made using tissue stored identically and therefore sample storage was not a confound in our studies. In our studies using qPCR and expression microarrays, data was compared using parametric statistics (t-tests or one-way ANOVAs) and, for qPCR, the validity of primer designs were confirmed by the supplier and therefore this should not have been a significant variable. The number of cycles required to give a measure of levels of RNA did not vary significantly between reference genes and our genes of interest and therefore reagents would not have been exhausted because we were trying to amplify genes with low copy number [7]. In conclusion there is no methodological variable that explains the different outcomes we have obtained measuring levels of RNA using qPCR or expression microarrays.
One final methodological consideration is how the high-throughput technologies and qPCR interact with RNA. Expression microarrays we use in our studies on the molecular pathology of psychiatric disorders in BA 10 [10,25] use 20mer probes to hybridize RNA for subsequent quantification [26]. RNA-seq uses next- generation sequencing (NGS) to quantify levels of RNA in a biological sample [27] whereas qPCR measures levels of an amplicon of cDNA sequence between two hybridised primers [28]. Significantly, total number of coding genes in the human genome is not markedly higher than those in much simpler eukaryotes but the number of variants produced during the transcription of both coding and non-coding RNA is higher in more complex organisms [29]. Therefore, differences in results from the use of expression microarrays and qPCR could be due to the ability to quantify different transcriptional variants. This is relevant to schizophrenia because differential expression of different gene variants has been suggested to be important in the molecular pathology of the disorder [30]. Notably, RNA-seq should have the advantage of providing data on all transcriptional variants [31].
Conclusion
In conclusion, whilst it has been a common practice to use qPCR data on a few genes to validate studies using high-throughput transcriptomic screening in the brain and blood from people with schizophrenia [32,33], data from this study shows that if we had used that approach to validate our study of changes in levels of coding and non-coding RNA in BA 10 [10] we would have concluded the expression microarray study was not valid. However, our follow up studies showing changes in levels of AKTIP and TUBA1B protein in BA 10 from people with schizophrenia [13,16] argues our transcriptomic study is pointing to both changes in transcription and translation in BA 10 that could be involved in the molecular pathology of schizophrenia. Given the potential importance of BA 10 in the pathophysiology of schizophrenia [11], not continuing to understand the full biological consequences of changes in the human transcriptome in that brain region due to our qPCR data could have been a significant error. Importantly, there is no clear methodological factor, other than a potential difference in being able to detect coding and non-coding RNA variants, which could account for the different outcomes we have from measuring RNA in BA 10 from controls and people with schizophrenia. Notably, the issue of different sensitivities to detecting transcriptional variants can be overcome by repeating our studies in BA 10 using RNA-seq that would provide data on RNA variants [31]. However, RNA-seq is a complex methodology and it may not be giving consistent results when used to measure levels of RNA [34]. Finally, whilst the study of the transcriptome is giving invaluable new insights into the molecular pathology of schizophrenia [35], it must be acknowledged that the functioning of any tissue is governed by levels and activities of proteins [12] and is therefore not necessarily reflected in changes in levels of RNA. In this respect, this study confirms that the complex regulation of gene translation by a cell [12] means that changes at the level of the transcriptome do not necessarily reflect changes at the level of the proteome.
References
- Osimo EF, Beck K, Reis Marques T, Howes OD. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol Psychiatry. 2019; 24: 549-61.
- Gibbons A, Sundram S, Dean B. Changes in Non-Coding RNA in Depression and Bipolar Disorder: Can They Be Used as Diagnostic or Theranostic Biomarkers? Noncoding RNA. 2020; 6.
- Horvath S, Mirnics K. Schizophrenia as a disorder of molecular pathways. Biol Psychiatr. 2015; 77: 22-8.
- Hoffman GE, Bendl J, Voloudakis G, Montgomery KS, Sloofman L, Wang YC, et al. CommonMind Consortium provides transcriptomic and epigenomic data for Schizophrenia and Bipolar Disorder. Scientific Data. 2019; 6: 180.
- Freeman WM, Walker SJ, Vrana KE. Quantitative RT-PCR: pitfalls and potential. Biotechniques. 1999; 26: 112-22.
- Kozera B, Rapacz M. Reference genes in real-time PCR. J Appl Genet. 2013; 54: 391-406.
- Korenková V, Scott J, Novosadová V, Jindrichová M, Langerová L, Švec D, et al. Pre- amplification in the context of high-throughput qPCR gene expression experiment. BMC Mol Biol. 2015; 16: 5.
- Dean B, Udawela M, Scarr E. Validating reference genes using minimally transformed qpcr data: findings in human cortex and outcomes in schizophrenia. BMC Psychiatry. 2016; 16: 154.
- Silberberg G, Baruch K, Navon R. Detection of stable reference genes for real-time PCR analysis in schizophrenia and bipolar disorder. Anal Biochem. 2009; 391: 91-7.
- Scarr E, Udawela M, Dean B. Changed frontal pole gene expression suggest altered interplay between neurotransmitter, developmental, and inflammatory pathways in schizophrenia. NPJ Schizophr. 2018; 4: 4.
- Snelleksz M, Rossell SL, Gibbons A, Nithianantharajah J, Dean B. Evidence that the frontal pole has a significant role in the pathophysiology of schizophrenia. Psychiatry Res. 2022; 317: 114850.
- Nelson DL, Cox MM. Lehninger Principles of Biochemistry. New York: W.H. Freeman and Company; 2005.
- Snelleksz M, Dean B. Lower levels of tubulin alpha 1b in the frontal pole in schizophrenia supports a role for changed cytoskeletal dynamics in the aetiology of the disorder. Psychiatry Res. 2021; 303: 114096.
- Greenwood TA, Shutes-David A, Tsuang DW. Endophenotypes in Schizophrenia: Digging Deeper to Identify Genetic Mechanisms. Journal of psychiatry and brain science. 2019; 4.
- Scarr E, Cowie TF, Kanellakis S, Sundram S, Pantelis C, Dean B. Decreased cortical muscarinic receptors define a subgroup of subjects with schizophrenia. Mol Psychiatr. 2009; 14: 1017-23.
- Snelleksz M, Dean B. ACNP 61st Annual Meeting: Poster Abstracts P271-P540. Neuropsychopharmacol. 2022; 47: 340.
- Dean B, Pavey G, Chai SY, Mendelsohn FAO. The localisation and quantification of molecular changes in the human brain using in situ radioligand binding and autoradiography. In: Dean B, Kleinman JE, Hyde TM, editors. Using CNS tissue in psychiatric research: A practical guide. Amsterdam: Harwood Academic Press. 1999; 67-83.
- Ferrer I, Armstrong J, Capellari S, Parchi P, Arzberger T, Bell J, et al. Effects of formalin fixation, paraffin embedding, and time of storage on DNA preservation in brain tissue: a BrainNet Europe study. Brain Pathol. 2007; 17: 297-303.
- Kingsbury AE, Foster OJ, Nisbet AP, Cairns N, Bray L, Eve DJ, et al. Tissue pH as an indicator of mRNA preservation in human post-mortem brain. Brain Res Mol Brain Res. 1995; 28: 311-8.
- Roberts SB, Hill CA, Dean B, Keks NA, Opeskin K, Copolov DL. Confirmation of the diagnosis of schizophrenia after death using DSM-IV: a Victorian experience. Aust N Z J Psychiatry. 1998; 32: 73-6.
- Hill C, Keks N, Roberts S, Opeskin K, Dean B, MacKinnon A, et al. Problem of diagnosis in postmortem brain studies of schizophrenia. Am J Psychiatry. 1996; 153: 533-7.
- D’Agostino RB, Belanger A, D’Agostino RB. A Suggestion for Using Powerful and Informative Tests of Normality. The American Statistician. 1990; 44: 316-21.
- Dean B. Irreproducible data: Problems and solutions for psychiatry. Aust NZ J Psychiatry. 2018; 52: 10-2.
- Lipska BK, ep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, et al. Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol Psychiatr. 2006; 60: 650-8.
- Scarr E, Udawela M, Dean B. Changed cortical risk gene expression in major depression and shared changes in cortical gene expression between major depression and bipolar disorders. Aust NZ J Psychiatry. 2019; 53: 1189-98.
- Chambers RC. Gene expression profiling: good housekeeping and a clean message. Thorax. 2002; 57: 754-6.
- Kukurba KR, Montgomery SB. RNA Sequencing and Analysis. Cold Spring Harb Protoc. 2015; 2015: 951-69.
- Smith CJ, Osborn AM. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol Ecol. 2009; 67: 6-20.
- Dhamija S, Menon MB. Non-coding transcript variants of protein-coding genes - what are they good for? RNA Biol. 2018; 15: 1025-31.
- Kaalund SS, Newburn EN, Ye T, Tao R, Li C, Deep-Soboslay A, et al. Contrasting changes in DRD1 and DRD2 splice variant expression in schizophrenia and affective disorders, and associations with SNPs in postmortem brain. Mol Psychiatry. 2014; 19: 1258-66.
- Cmero M, Schmidt B, Majewski IJ, Ekert PG, Oshlack A, Davidson NM. MINTIE: identifying novel structural and splice variants in transcriptomes using RNA-seq data. Genome Biol. 2021; 22: 296.
- Narayan S, Tang B, Head SR, Gilmartin TJ, Sutcliffe JG, Dean B, et al. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res. 2008; 1239: 235-48.
- Cattane N, Minelli A, Milanesi E, Maj C, Bignotti S, Bortolomasi M, et al. Altered gene expression in schizophrenia: findings from transcriptional signatures in fibroblasts and blood. PLoS One. 2015; 10: e0116686.
- Simoneau J, Dumontier S, Gosselin R, Scott MS. Current RNA-seq methodology reporting limits reproducibility. Briefings in Bioinformatics. 2019; 22: 140-5.
- Nakamura T, Takata A. The molecular pathology of schizophrenia: an overview of existing knowledge and new directions for future research. Mol Psychiatr. 2023; 28: 1868-89.