
Research Article
J Schizophr Res. 2021; 7(1): 1038.
The Schizophrenia Variant V1282F in SCN2A Causes Functional Impairment of NaV1.2
Kohlnhofer B, Liu Y, Woodruff G, Lovenberg T, Bonaventure P and Harrington AW*
Neuroscience Discovery, Janssen Pharmaceutical Companies of Johnson & Johnson, USA
*Corresponding author: Anthony W Harrington, Neuroscience Discovery, Janssen Pharmaceutical Companies of Johnson & Johnson, 3210 Merryfield Row, San Diego, CA 92121, USA
Received: March 17, 2021; Accepted: April 01, 2021; Published: April 08, 2021
Abstract
Neuropsychiatric disorders such as schizophrenia are challenging to treat due to the biological complexity of the disease and the lack of knowledge of the underlying pathophysiology. Whole exome and genome sequencing studies have identified disease-linked rare variants in patients with large effect size. Here, we functionally characterize the schizophrenia linked variant V1282F in SCN2A, encoding the sodium channel Nav1.2. This variant was introduced into isogenic lines of hiPSCs using CRISPR/CAS9 genome editing tools. hiPSCs were then differentiated into cortical neurons to understand how the variant and gene may be contributing to disease. We observed a significant (~25%) decrease in sodium current in the V1282F neurons compared to control neurons, suggesting the mutation is causing a loss-of-channel function. These results were supported by recordings in recombinant cells overexpressing either the mutant or wildtype Nav1.2, with the mutant channel having significantly (~75%) lower current amplitude than wildtype. We hypothesize that this phenotype may contribute to disease either through the direct loss of neuronal activity or through subsequent abnormal neurodevelopment.
Introduction
Elucidating novel therapeutic targets for schizophrenia has been difficult due to the lack of knowledge of the fundamental biology and pathophysiology of this complex disease. Additionally, there is poor predictive validity in current pre-clinical models. Factoring into these problems is the lack of human model systems and our poor understanding of the disease genetics. Genetics are a major contributor to schizophrenia, as the heritability of schizophrenia is estimated to be 79% from twin studies [1]. With the recent advances in genome sequencing, we now have insight into genes that may be contributing to schizophrenia and that have the potential to be novel pharmaceutical targets. Genome Wide Association Studies (GWAS) and whole genome or exome sequencing efforts have identified variants in schizophrenia patients, presumed to be disease causing but it is not clear what the functional effects of these variants are, if any. Most of the GWAS-identified common variants are within non-coding regions, so the field resorts to investigating rare coding variants identified through whole genome or exome sequencing. To transition from the bench to the bedside, it is necessary to first understand how individual patient risk variants affect gene or gene product function and how this contributes to disease.
Many of the genes that contain variants for schizophrenia overlap with those that contain variants for Autism Spectrum Disorder (ASD) and Intellectual Disability (ID) [2]. The gene that harbors the greatest combined number of variants identified in ASD, ID, and schizophrenia patient populations is the voltage-gated Sodium Channel Alpha Subunit 2 (SCN2A). SCN2A encodes for the sodium channel Nav1.2, which is critical for the initiation and propagation of action potentials [3]. In fact, in excess of a hundred unique de novo mutations in SCN2A have been identified and with hundreds of children born each year having mutations in SCN2A [4,5]. These variants have been identified through whole genome or exome sequencing studies and although extremely rare, they are expected to have high penetrance. SCN2A variants have also been identified in patients with Infantile Epileptic Encephalopathy (IEE) and Benign Infantile Familial Seizures (BIFS), where the contribution of variants to disease is better understood and those variants that have been functionally analyzed have been found to be gain-of-function [6-11]. Through bioinformatic modeling, multiple SCN2A variants identified in neuropsychiatric patients were predicted to be loss-of-function and recently a few have been functionally evaluated using overexpression studies in HEK293 cells. Eleven unique ASD variants and three unique ID variants were found to either functionally inactive or inhibit channel function [12,13]. In mice, SCN2A knockout is embryonic lethal and multiple groups have found that SCN2A+/- mice display autistic and schizophrenia-like phenotypes [14-16].
While schizophrenia-linked mutations in SCN2A are less common, mutations have been identified through whole genome or exome sequencing, including one that leads to loss of a splice site (c.2150 A>G), one that generates a nonsense mutation (E169X), and one that creates a missense mutation (V1282F) [2,17]. However, none of them have been functionally investigated [2,17-19]. To date, there are no reports on the functionality or effect of SCN2A variants associated with schizophrenia. In this study, we investigated one coding variant, V1282F (Chr2: 166226804, (G/T)), located in the third transmembrane segment of the third domain. This is a missense variant that has been identified in two unrelated patients and is absent in the control dataset [17]. To investigate this variant in a biologically relevant system, we used human induced pluripotent stem cell (hiPSC) derived cortical neurons and introduced the variant using CRISPR/CAS9 genome editing tools to analyze the variant under endogenous expression levels. We found that the V1282F variant causes a significant decrease in Na+ channel current density. V1282F mutant channels exogenously expressed in HEK293 cells further showed lower current amplitudes than Wildtype (WT) channels, consistent with the findings in hiPSC-derived neurons and suggesting a loss-of-channel function associated with the V1282F mutant. To our knowledge, this is the first report of the functional impact of a SCN2A variant identified in a schizophrenia patient.
Methods
hiPSC maintenance and genome editing
The healthy control hiPSC line GM25430 (CVB) was obtained from the Coriell Institute (Camden, NJ). All hiPSC lines were maintained in mTeSR on vitronectin coated plates passaged using ReLeSR (Stem Cell Technologies, Vancouver, Canada). To generate SCN2AWT/V1282F and SCN2AV1282F/V1282F hiPSC lines, a guide RNA (gcatactcacATCAACAATC) was designed to target exon 19 near chromosome position: 166226804(G/T) using the guide design tool from crispr.mit.edu and a SSODN donor template was designed using 50bp homology arms incorporating the (G/T) variant (G T T T T C A A G T G T A T T T T A C C A A T G C C T G G T G C T G G C T A G A C T T C C T G A T T (T) T T G A T G T G A G T A T G C T G C A C T T T G C T G C T T T A T T C A T T G G C A T A T A T G T). Genome editing was performed using the Alt-R-CRISPR system (IDT, Coralville, IA). Alt-R S.p. HiFi Cas9 Nuclease V3, Alt-R CRISPR-Cas9 crRNA, Alt-R CRISPR-Cas9 tracrRNA (ATTO 550), and SSODN were nucleofected into hiPSCs using the P3 Primary Cell 4D-Nucleofector X kit and the Amaxa 4D-Nucleofector X Unit (Lonza, Basel, Switzerland) setting CB-150. 24 hours post-nucleofection ATTO 550+ single cells were FACS sorted and plated. Isogenic clones were screened for the variant by amplifying the DNA using Polymerase Chain Reaction (PCR) and primers flanking the variant (Primer F: aggagttcctgcaaatgagttaccc and Primer R: aatgttttgaggcatcctctcactg) followed by Sanger sequencing.
Neural precursor cell differentiation and neuron differentiation
hiPSCs were differentiated into Neural Precursor Cells (NPCs) over the course of three weeks using the STEMdiff Neural Induction Media + SMADi Kit and protocol (STEMCELL Technologies) on 0.01% Poly-L-ornithine and Laminin (ThermoFisher, Waltham, MA) coated plates. NPCs were cultured in neural maintenance media: 0.5X DMEM/F12 + Glutamax, Neurobasal , 0.5X N2 supplement, 0.5X B27 Supplement , 0.5X Glutamax, 0.5X IT-S, 0.25X 2-Mercaptoethanol (Gibco-ThermoFisher, Waltham, MA), Pen- Strep (Corning, Corning, NY) and NEAA (Hyclone Laboratories Inc- ThermoFisher, Waltham, MA), and 20ng/ml FGF2 (R&D Systems, Minneapolis, MN). NPCs were differentiated into neurons by withdrawing FGF2 once confluent. After three weeks, cells were replated for downstream assays and 24 hours later treated with 1uM cytosine arabinoside (ARA-C) (Sigma, St. Louis, MO) for 72 hours to eliminate proliferating cells. After ARA-C treatment the media was switched to neuronal maturation media containing Brain Phys (STEMCELL Technologies), 1X PenStrep, 1X N2 supplement, 1X B27 Supplement, 20ng/uL BDNF (Peprotech, Rocky Hill, NJ), 20ng/ uL GDNF (Peprotech), 1mM cAMP (Sigma, St. Louis, MO), 1ug/ mL Laminin, and 200nM Ascorbic Acid (Sigma). Neurons were matured for an additional month with weekly half media changes. All assays were performed at eight weeks after FGF2 withdrawal unless indicated.
RNA isolation, cDNA synthesis and qRT-PCR
RNA was isolated from hiPSCs or two-month neuronal cultures using a RNeasy Mini Kit and samples were DNase treated on column (Qiagen, Hilden, Germany). cDNA was synthesized using SuperScript IV First-Strand Synthesis System (Invitrogen, Carlsbad, CA) and qRT-PCR was performed using TaqMan assays (ThermoFisher, Waltham, MA) and analyzed on a QuantStudio Real Time PCR System (Applied Biosystems, Foster City, CA). TaqMan assays: OCT4: Hs00999634_gH, SCN2A: Hs01109871_m1, SCN3A: Hs00366902_m1, SCN8A: Hs00274075_m1, and GAPDH: Hs02786624_g1. Data were normalized to GAPDH expression levels. Data were plotted as mean ±SEM. Statistical analysis was performed using a one-way ANOVA and results were considered significant at p-value <0.05 (GraphPad Prism).
Immunofluorescence
NPCs were fixed in 4% Paraformaldehyde (PFA) for five minutes and two-month neuronal cultures were fixed in with 4% PFA for 30 minutes. Samples were permeabilized and blocked in 0.5% PB-Triton and 5% BSA for one hour. Primary antibodies: anti- Nav1.2, Cat#: ASC-002 (Alomone Labs, Jerusalem, Israel); MAP2, Cat#: AB5392 (Abcam, Cambridge, United Kingdom); SYP, Cat#: AB14692 (Abcam); SOX2, Cat#: AB97959 (Abcam); NES, Cat#: ab22035 (Abcam) were incubated at 4°C overnight and secondary antibodies were incubated at room temperature for one hour. Images were captured on an Opera Phenix high-content imager at 20x magnification (PerkinElmer, Waltham, MA) and analyzed using their Columbus Image Data Storage and Analysis System software. Synapse density was quantified by having the software find puncta in the area surrounding the neurites. Images were analyzed from two lines from each genotype, three differentiations, with at least six wells with five images from each well. Data were plotted as mean ±SEM. Statistical analysis was performed using a one-way ANOVA and results were considered significant at p-value <0.05 (GraphPad Prism).
SCN2A pulldown and simple western blot- protein simple wes/jess platform
Nav1.2 membrane protein was isolated from two-month neurons by preincubation of the neurons with the 100ug/ml leupeptin (Sigma) for 30 minutes at 37°C, biotinylation of membrane proteins in 1.5mg/ ml sulfo-NHS-SS-biotin (Sigma) for 20 minutes at 4°C rotating, cell lysis, and pull-down of the biotinylated protein using neutravidinagarose beads (Pierce, Waltham, MA). Both membrane Nav1.2 and total Nav1.2 samples were probed using Nav1.2 ASC-002 (Alomone Labs) at 1:500 via western using the Jess High Molecular Weight 66- 440 kDa Separation Module (Protein Simple, San Jose, CA). Total Nav1.2 samples were normalized to total protein using the Protein Simple- Total Protein Assay (Protein Simple) and membrane Nav1.2 was normalized to input Nav1.2 protein. Data were plotted as mean ±SEM. Statistical analysis was performed using a one-way ANOVA and results were considered significant at p-value <0.05 (GraphPad Prism).
Electrophysiology
For manual patch clamp recording, neurons were cultured at a density of 3×105 cells on 15mm glass coverslips placed in a chamber on the stage of an inverted microscope. Upon formation of the whole-cell conformation, the cell was constantly perfused with an extracellular solution containing (in mM): 74.5 NaCl, 74.5 choline Cl, 2.5 BaCl2, 2 KCl, 1 MgCl2, 5 glucose, 10 HEPES, 0.3 CdCl2, 3 4-aminopyridine, pH 7.4, 310 mOsm/L. The pipette electrode was filled with an intracellular solution containing (in mM): 115 Cs methanesulfonate, 20 CsCl, 3 NaCl, 4 MgATP, 0.3 Na2GTP, 10 EGTA and 20 HEPES, pH 7.2, 290 mOsm/L. Currents were measured by whole-cell patch clamp using an Axopatch 200B amplifier and pClamp 11 software (Molecular Devices, San Jose, CA), digitized at 20 kHz and lowpass filtered at 5 kHz. Series resistance was 75% compensated. Currents were leak subtracted using a P/4 protocol.
For automated Syncropatch electrophysiology (Nanion, Germany), cDNA of the a subunit of the human Nav1.2 WT and V1282F mutant was transfected into HEK293 cells (76 μg each per T75 flask). Transfected cells were kept at 37°C for the first day and moved to 30°C for the second day before experiment. Single-hole chips with medium resistance were used. The extracellular solution contained (in mM): 149 NaCl, 2.8 CaCl2, 4 KCl, 1 MgCl2, 5 glucose, 10 HEPES, pH 7.4, 310 mOsm/L. The intracellular solution contained (in mM): 110 CsF, 10 NaCl, 10 KCl, 10 EGTA, 10 HEPES, pH7.2, 280 mOsm/L. Currents were digitized at 20 kHz and lowpass filtered at 5 kHz. Series resistance was automatically compensated. Currents were leak subtracted.
The voltage protocol was the same for both manual and automated patch clamp. From a holding potential of -100 mV, a 100-ms preconditioning pulse ranging from -100 mV to +40 mV (in 10 mV increments) was followed by a 10 ms test pulse to 0 mV and subsequent return to -100 mV. The time interval between preconditioning pulses was five seconds. The peak current amplitudes during the preconditioning pulses were used to calculate the voltage dependence of activation (I-V/G-V). The peak current amplitudes during the test pulses were used to calculate the current density and voltage dependence of inactivation. All electrophysiological recordings were performed at room temperature.
Electrophysiology data analysis
V1/2, the voltage at which the conductance is 50% of the maximum value, was obtained by fitting the conductance-voltage (G-V) relationship to a Boltzmann function. Kinetic parameters were obtained by fitting the data with a single exponential function. Data fitting and statistical analyses (two-tailed Student’s t-test or two-way ANOVA as indicated) were performed using Origin (Northampton, MA, USA). Results were considered statistically significant at p-value <0.05. Experimental data are reported as mean ±SEM, which result from independent measurements on n different cells.
Results
Generation and characterization of isogenic SCN2A V1282F hiPSCs and neurons
To investigate whether the SCN2AV1282F/V1282F variant has any effect in hiPSC-derived cortical neurons, we used CRISPR/CAS9 genome editing tools to introduce the (G/T) variant into control hiPSCs. The (G/T) variant is in exon 19 and results in the substitution of a valine residue for a phenylalanine residue in the third segment of the third transmembrane domain in Nav1.2 (Figure 1A). This variant has been identified in two schizophrenia patients, is absent from the control EVS population and statistics suggest this is a nonrandom association by the Fischer’s exact test, 2-tailed p-value=0.034 [17]. However, the functional effects of V1282F are unknown.
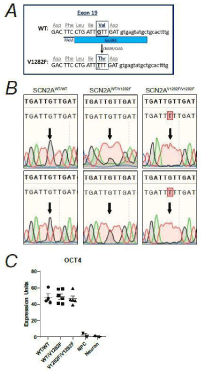
Figure 1: Generation and characterization of SCN2AV1282F/V1282F hiPSC lines.
(A) A schematic of the CRISPR/CAS9 targeting strategy introducing the
V1282F variant into exon 19 of SCN2A. (B) Chromatographs of the hiPSC
lines generated for each genotype obtained by Sanger sequencing.
(C) Gene expression of the pluripotency marker OCT4 in hiPSC lines,
normalized to GAPDH. Data from iPSCs was derived from two independent
lines/genotype (n= 2 WT/WT, n=2 WT/V1282F, n=2 V1282F/V1282F), one
wildtype NPC sample, and one wildtype two-month old neuron sample. All
samples were run in duplicate.
We generated two hiPSC lines for each genotype: wildtype homozygous (G/G) SCN2AWT/WT, heterozygous (G/T) SCN2AWT/ V1282F, and V1282F homozygous (T/T) SCN2AV1282F/V1282F (Figure 2B). The hiPSC lines were confirmed to express the pluripotent stem cell transcript OCT4 (Figure 1C), which confirms that the genome editing process did not affect pluripotency of the resulting isogenic clones. All hiPSCs were differentiated into Neural Precursor Cells (NPCs) and probed for the NPC proteins SRY-box transcription factor 2 (SOX2) and Nestin to confirm NPC identity (Figure 2A). NPCs were differentiated down the cortical neuronal lineage and cultured for two months. We first characterized the control and variant neurons by staining for the Microtubule Associated Protein 2 (MAP2) and the presynaptic protein Synaptophysin (SYP) and analyzed neurite length and synapse density. We observed no significant differences in neurite length or synapse density between the three genotypes (Figure 2B and 2C). Together these data suggest that the variant does not affect neuronal differentiation, neurite elongation or synapse formation at this early stage in vitro.
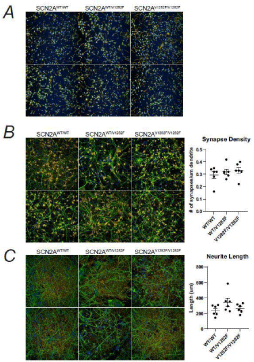
Figure 2: SCN2A V1282F variant does not affect synapse number or neurite
length.
(A) Immunofluorescent staining for the NPC markers NESTIN and SOX2 in
NPCs.
(B) Immunofluorescent staining for SCN2A and the neuronal marker MAP2 in
two-month cortical neurons and quantification of neurite length.
(C) Immunofluorescent staining for the neuronal marker MAP2 and the
pre-synaptic marker SYP in two-month cortical neurons and quantification
of synapse number per micron dendrite. Data was derived from two
independent lines/genotype and at least three differentiations/line, at least six
wells/differentiation and five images/well were analyzed.
Sodium current density in SCN2AV1282F/V1282F neurons is decreased
To determine if the missense variant alters Na+ channel properties in these neurons, we performed whole-cell manual patch clamp studies on SCN2AWT/WT and SCN2AV1282F/V1282F neurons. No significant differences (p>0.05) in the kinetics of channel inactivation (Figure 3A and 3B) or the voltage dependence of channel activation and inactivation (Figure 3C) were observed between SCN2AWT/ WT and SCN2AV1282F/V1282F neurons. However, the mean Na+ current density in SCN2AV1282F/V1282F neurons (881.5±73.8, n=45) was significantly (p=0.017) smaller (by ~25%) than that in SCN2AWT/WT neurons (1162.7±88.5, n=42) (Figure 3A and 3D), suggesting that the SCN2AV1282F/V1282F variant may alter Nav1.2 channel function (e.g. channel open probability, single channel conductance, etc.) or the number of functional Nav1.2 and/or other Na+ channels at the cell surface.
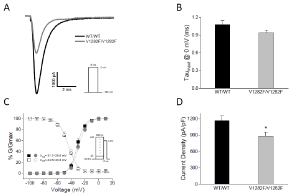
Figure 3: Na+ current density is decreased in SCN2AV1282F/V1282F neurons.
(A) Representative Na+ current traces from SCN2AWT/WT (black) and
SCN2AV1282F/V1282F (gray) neurons.
(B) The mean time constant of Na+ channel inactivation (at 0 mV) for
SCN2AV1282F/V1282F neurons (n=42) is not significantly different (p>0.05; twotailed
Student’s t-test) from that for SCN2AWT/WT neurons (n=40).
(C) The voltage dependence of channel activation and inactivation for
SCN2AWT/WT (solid and outlined squares, respectively; n=34) and SCN2AV1282F/
V1282F (solid and outlined circles, respectively; n=34) neurons is not significantly
different (p>0.05; two-way ANOVA).
(D) The mean Na+ current density of SCN2AWT/WT is significantly different from
that of SCN2AV1282F/V1282F neurons (p=0.017; two-tailed Student’s t-test).
V1282F mutant channels have decreased whole-cell current amplitude in HEK293 cells
Since this is a native model system in which other sodium channels are likely contributing to the overall sodium current, we cannot isolate the sole contribution of Nav1.2 from the overall current. To better understand the functional effect of the variant on Nav1.2 channel specifically, we recorded from HEK293 cells transiently transfected with either Nav1.2 WT or V1282F mutant channels using the Syncropatch automated patch clamp instrument. The V1282F channel was functional but when compared to the WT channel, the mean amplitude of the whole-cell current in cells expressing V1282F mutant channels was significantly smaller (by ~75%; p<0.0001) (Figure 4A). In addition, V1282F produced a relatively small (~3.5 mV), but statistically significant (p<0.001), hyperpolarizing shift in the voltage dependence of channel inactivation (Figure 4B). The voltage dependence of channel activation, on the other hand, was not significantly (p>0.05) different between WT and V1282F channels (Figure 4C).
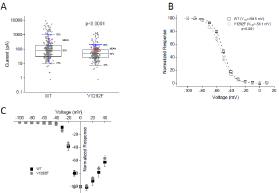
Figure 4: Functional characterization of human Nav1.2 WT and V1282F
mutant channels in HEK293 cells by Syncropatch electrophysiology.
(A) Box plot of the current amplitude in cells transiently transfected with WT
and V1282F. The mean amplitude of the V1282F current (119.2±14.7 pA;
n=226) is significantly smaller (p<0.0001; two-tailed Student’s t-test) than that
of the WT current (456.8±68.9 pA; n=264).
(B) Voltage dependence of WT (outlined squares) and V1282F (outlined
circles) channel inactivation. The V1/2 of V1282F channels (-54.1±0.2 mV;
n=19) is significantly different (p<0.001; two-way ANOVA) from that of WT
channels (-50.5±0.1 mV; n=22).
(C) The current-voltage relationship (normalized to the current amplitude at 0
mV) of V1282F channels (solid circles; n=19) is not significantly different from
that of WT channels (solid squares; n=22) (p>0.05).
SCN2A V1282F variant does not alter Nav1.2 membrane expression or localization
A decrease in sodium current could be caused by a decrease in SCN2A expression level or a decrease in the number of Nav1.2 channels present at the plasma membrane. To investigate these theories, we measured SCN2A gene expression, protein and membrane localization levels in SCN2AWT/WT, SCN2AWT/V1282F, and SCN2AV1282F/ V1282 neurons. We found that SCN2A gene expression was unaltered between the different genotypes (Figure 5A). Interestingly, we observed a 28% increase in total Nav1.2 protein levels in SCN2AV1282F/ V1282F neurons compared to SCN2AWT/WT neurons (Figure 5B). Notably, the SCN2AWT/V1282F neurons had a level of protein in between the wildtype and homozygous variant neurons. No change in plasma membrane Nav1.2 levels was observed (Figure 5C). Together these data demonstrate that the V1282F variant does not inhibit SCN2A expression or SCN2A localization but does slightly increase SCN2A protein levels.
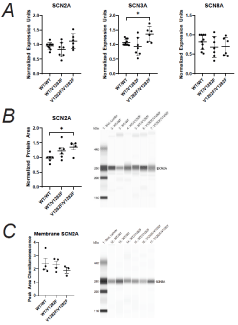
Figure 5: Expression of Na+ channels in SCN2AWT/WT, SCN2AWT/V1282F and
SCN2AV1282F/V1282F neurons.
(A) Quantification of SCN2A, SCN3A, and SCN8A gene expression in
SCN2AWT/WT, SCN2AWT/V1282F SCN2AV1282F/V1282F neurons, normalized to
GAPDH.
(B) Quantification of total Nav1.2 levels normalized to total protein.
*p-value=<0.05.
(C) Quantification of Nav1.2 membrane protein normalized to total SCN2A
input. All assays were performed in two-month old cortical neurons from at
least two independent lines for each genotype and at least two differentiations
per line were analyzed.
SCN3A and SCN8A sodium channel gene expression
In addition to Nav1.2, other members of the voltage-gated sodium channel family, notably Nav1.3 (SCN3A) and Nav1.6 (SCN8A), are also significantly expressed in these neurons. We next asked if the V1282F variant might alter the expression of SCN3A or SCN8A, which could also result in changes in Na+ current density via compensatory mechanisms. To address this question, we performed qRT-PCR to quantify the mRNA transcript levels. We identified a 20% increase in SCN3A levels in SCN2AV1282F/V1282F neurons, but no change in SCN2AWT/V1282F neurons when compared to the SCN2AWT/WT neurons. On the other hand, SCN8A levels were not significantly different for the three genotypes (Figure 5A).
Discussion
Nav1.2 variants associated with IEE and BIFS are thought to result in a gain-of-function, whereas those implicated in ASD and ID are thought to result in a loss-of-function. Several Nav1.2 variants have also been identified as linked to schizophrenia. However, none of them have been functionally investigated. In this report, we studied the effect of the schizophrenia associated SCN2A variant, V1282F, in a native neuronal environment. We engineered the mutation in isogenic hiPSC-derived cortical neurons and assessed Na+ channel activity as well as other functional and biochemical endpoints and consequences. To our knowledge, this is the first SCN2A variant functionally modeled in hiPSC-derived neurons.
We found that hiPSC-derived cortical neurons containing the V1282F missense mutation in SCN2A have decreased Na+ current density. Consistent with this, V1282F mutant channel expressed in HEK293 cells produce smaller macroscopic currents than WT channels. As such, our results indicate that V1282F results in lossof- function phenotypes both in hiPSC-derived neurons and in a heterologous system. Two ASD missense mutations (D82G and T1429M) assessed in HEK293 cells have also been found to have decreased peak currents, supporting the hypothesis that SCN2A schizophrenia variants functionally align with ASD variants over IEE and BIFS variants [12]. The observed increase in SCN3A gene expression in hiPSC-derived neurons may suggest a compensatory mechanism for a decrease in SCN2A function. While we saw a significant increase in SCN2A protein in these neurons, we did not observe a significant difference in SCN2A mRNA. This increase in protein could be rationalized by the slight but not significant increase in SCN2A mRNA and/or the possibility that the SCN2A V1282F protein is more stable. Potentially, the increase in SCN2A protein could also be due to a feedback mechanism to compensate for the decrease in Na+ current, which suggest the neurons are trying to increase channel number to compensate for the functionally impaired protein.
Additionally, our interest lies in how a 25% decrease in current density may affect overall network activity. With the development of new high-density multi-electrode arrays, it is now possible to address this question in future studies. Deneault et al. engineered a SCN2A knockout in iPSC-derived neurons and found that SCN2A-/- neurons had decreased sEPSC frequency but no change in amplitude [20]. Moreover, they used multi-electrode arrays to analyze network activity and found SCN2A-/- neurons had a decreased mean firing rate and decreased burst frequency [20]. These findings suggest that SCN2AV1282F/V1282F neurons may also have altered network activity.
While mutations in SCN2A do have various effects on Na+ channel function, as previously shown by others and here by us, how these changes in Na+ channel activity lead to a pathological mechanism of disease is not well understood. Neuronal electrical activity is required for proper development of the nervous system, specifically in neurite outgrowth, dendritic growth, axon pathfinding and synapse formation [21,22]. Moreover, expression of voltage-gated sodium channels is tightly coordinated during development. As SCN2A knockout mice are perinatal lethal, the contribution of SCN2A is vital [14]. SCN2A is expressed along the length of the axon in early development before it is eventually replaced by SCN8A. At this point, SCN2A becomes localized to the axon initial segment and nodes of Ranvier [23-25]. A decrease in the effective function of SCN2A during these early phases of neurodevelopment could have a lasting impact, for example, later in adolescence and early adulthood when the clinical symptoms of schizophrenia manifest.
The hiPSC-derived cortical neuron cultures used for our studies represent early embryonic cortical development [26]. We propose that we may be modeling the early developmental stages of disease with this in vitro culture system, thus the significant difference in activity of the channel in this system likely translates to the early in vivo situation. Since our neuronal cultures are more fetal-like than adult, this could be why we did not observe decreased synapse density in our SCN2AV1282F/V1282F neurons. However, other hiPSC disease modeling studies have reported decreased synapse density [27,28], so another possibility is simply that the V1282F variant does not result in reductions in synapse density. When SCN2A mutations linked with ASD were computationally modeled in a mature model system, where SCN8A protein is elevated and SCN2A protein becomes localized to the axon initial segment, only modest phenotypes were observed [12]. These data suggest that those variants played a more active developmental role in the maturation of the brain, which is important for how we design therapeutics to treat disease. Future studies should elucidate whether SCN2A could be a potential therapeutic target, and if so, what the timing and mechanism of intervention would consist of.
The contribution of SCN2A variants to neuropsychiatric disease is gaining traction and our data here further reveal that a SCN2A schizophrenia risk variant may functionally contribute to disease by impairing Nav1.2 channel function and subsequent Na+ current density. This agrees with evidence from a clinical study that found that schizophrenia patients with poorer cognitive performance had lower levels of SCN2A expression, suggesting this gene may be implicated more widely in schizophrenia in general [18].
hiPSC disease modeling studies of rare variants associated with schizophrenia are an important step in understanding the underlying mechanism of disease. Moving forward, the knowledge gained from using these approaches will allow us to understand how certain mutations may lead to dysfunction severe enough to cause disease as well as to discover new therapeutics tailored to the specific dysfunction of that gene. Here we have demonstrated that a rare variant in SCN2A identified in two unrelated schizophrenia patients causes decreased Na+ current. Whether this information can be leveraged into a therapeutic strategy for schizophrenia patients harboring the V1282F mutation is a question for future studies.
References
- Hilker R, Helenius D, Fagerlund B, Skytthe A, Christensen K, Werge TM, et al. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biological Psychiatry. 2018; 83: 492-498.
- Fromer M, Pocklington A, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014; 506: 179-184.
- Li J, Cai T, Jiang Y, Chen H, He X, Chen C, Li X, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry. 2016; 21: 290-297.
- Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017; 140: 1316-1336.
- Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018; 41: 442-456.
- Liao Y, Deprez L, Maljevic S, Pitsch J, Claes L, Hristova D, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain. 2010; 133: 1403-1414.
- Misra SN, Kahlig KM, George AL. Impaired Nav1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia. 2008; 49: 1535-1545.
- Xu R, Thomas EA, Jenkins M, Gazina EV, Chiu C, Heron SE, et al. A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Molecular and Cellular Neuroscience. 2007; 35: 292-301.
- Lauxmann S, Boutry-Kryza N, Rivier C, Mueller S, Hedrich UBS, Maljevic S, et al. An SCN2A mutation in a family with infantile seizures from Madagascar reveals an increased subthreshold Na+ current. Epilepsia. 2013; 54: e117-e121.
- Ogiwara I, Ito K, Sawaishi Y, Osaka H, Mazaki E, Inoue I, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009; 73: 1046-1053.
- Schwarz N, Hahn A, Bast T, Muller S, Loffler H, Maljevic S, et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. Journal of Neurology. 2016; 263: 334-343.
- Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, Bender KJ. Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol Psychiatry. 2017; 82: 224-232.
- Begemann A, Acuna MA, Zweier M, Vincent M, Steindl K, Bachmann- Gagescu R, et al. Further corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes. Molecular Medicine. 2019; 25: 6.
- Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, et al. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha (II)-deficient mice. Biophys J. 2000; 78: 2878-2891.
- Tatsukawa T, Raveau M, Ogiwara I, Hattori S, Miyamoto H, Mazaki E, et al. SCN2A haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity. Mol Autism. 2019; 10: 15.
- Middleton SJ, Kneller EM, Chen S, Ogiwara I, Montal M, Yamakawa K, et al. Altered hippocampal replay is associated with memory impairment in mice heterozygous for the Scn2a gene. Nature Neuroscience. 2018; 21: 996-1003.
- Carroll LS, Woolf R, Ibrahim Y, Wiliiams HJ, Dwyer S, Walters J, et al. Mutation screening of SCN2A in schizophrenia and identification of a novel loss-of-function mutation. Psychiatr Genet. 2016; 26: 60-65.
- Dickinson D, Straub RE, Trampush JW, Gao Y, Feng N, Xie B, et al. Differential effects of common variants in SCN2A on general cognitive ability, brain physiology, and messenger RNA expression in schizophrenia cases and control individuals. JAMA Psychiatry. 2014; 71: 647-656.
- Suddaby JS, Silver J, So J. Understanding the schizophrenia phenotype in the first patient with the full SCN2A phenotypic spectrum. Psychiatric Genetics. 2019; 29: 91-94.
- Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, et al. Complete Disruption of Autism-Susceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Reports. 2018; 11: 1211-1225.
- Casagrande VA, Condo GJ. The effect of altered neuronal activity on the development of layers in the lateral geniculate nucleus. J Neurosci. 1988; 8: 395-416.
- Riccio RV, Matthews MA. The postnatal development of the rat primary visual cortex during optic nerve impulse blockade by intraocular tetrodotoxin: a quantitative electron microscopic analysis. Brain Res. 1985; 352: 55-68.
- Garrido JJ, Fernandes F, Giraud P, Mouret I, Pasqualini E, Fache MP, et al. Identification of an axonal determinant in the C-terminus of the sodium channel Nav1.2. The EMBO Journal. 2001; 20: 5950-5961.
- Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA. Differential Control of Clustering of the Sodium Channels Nav1.2 and Nav1.6 at Developing CNS Nodes of Ranvier. Neuron. 2001; 30: 105-119.
- Kole MHP, Stuart GJ. Signal Processing in the Axon Initial Segment. Neuron. 2012; 73: 235-247.
- Livesey MR, Magnani D, Hardingham GE, Chandran S, Wyllie DJA. Functional properties of in vitro excitatory cortical neurons derived from human pluripotent stem cells. J Physiol. 2016; 594: 6573-6582.
- Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2014; 515: 414-418.
- Brennand KJ, Gage FH. Modeling psychiatric disorders through reprogramming. Dis Model Mech. 2011; 5: 26-32.