
Research Article
J Stem Cells Res, Rev & Rep. 2014;1(3): 1015.
Mesenchymal Stem Cell Conditioned Media (MSC-CM) Suppress Wnt-3a and TGF-β1 - Induced Myofibroblastic Differentiation
Sohel Samad1*, Khondoker M Akram1, Nicholas R Forsyth1 and Monica A Spiteri1,2
1Institute for Science and Technology in Medicine, Keele University, UK
2Department of Respiratory Medicine, University Hospital of North Staffordshire, UK
*Corresponding author: Sohel Samad, Institute for Science and Technology in Medicine, Keele University, UK
Received: October 28, 2014; Accepted: November 25, 2014; Published: November 28, 2014
Abstract
Hypothesis: Idiopathic Pulmonary Fibrosis (IPF) remains an incurable fibrotic lung disease. A human Mesenchymal Stem Cell (MSC)-mediated regenerative approach has been proposed; MSC-mediated anti- fibrotic effects have been demonstrated in animal lung-fibrosis models. However the mechanism of action and effect on myofibroblastic differentiation are unknown. The Wnt family member, Wnt-3a, has been implicated as an inducer of myofibroblastic differentiation in fibroblast cell models. This study explores the influence of MSC secreted factors on Wnt-3a and TGF-β1-induced lung myofibroblastic differentiation.
Method: Human normal lung (CCD-8Lu) and IPF (LL29) fibroblasts were differentiated with Wnt-3a for 72-hours and TGF-β1 for 24-hours. MSCmediated differentiation inhibition was assessed by co-incubation of fibroblasts with MSC-CM and either Wnt-3a for 72-hours or TGF-β1 for 24-hours. TGF- β1-induced myofibroblastic differentiation reversal was explored with MSC-CM incubation for 24, 48 and 72-hours. Myofibroblast differentiation was assessed by immunocytochemical detection of α-smooth muscle actin expression
Results: Myofibroblastic differentiation following TGF-β1 treatment was achieved in 86.27±2.57% CCD-8Lu cells and 86.69±2.51% LL29 cells respectively. Similar, though reduced, levels of myofibroblastic differentiation were achieved in 52.9±0.20% CCD-8Lu and 55.60±5.90% LL29 cells respectively following Wnt-3a treatment.
In contrast, a percentage reduction in myofibroblastic differentiation was achieved in CCD-8Lu 31.40±1.44% and LL29 35.69±7.47% cells following exposure to TGF-β1 in the presence of MSC-CM versus TGF-β1 alone (p<0.001). Similarly, we observed a striking percentage reduction in myofibroblastic differentiation following co-incubation with Wnt-3a and MSC-CM versus Wnt- 3a alone (p<0.001); 80.76±3.64% of CCD-8Lu and 79.67±3.94% of LL29 cells.
A reversal of myofibroblastic differentiation was observed in TGF-β1- induced myofibroblasts following 72-hours administration of MSC-CM compared to serum-free culture media (p<0.001). Interestingly, we observed a MSCCM exposure duration effect on the total myofibroblast percentage present in both CCD8-Lu and LL29 cells; 81.70+0.43% and 73.26+0.70% respectively at 24-hours, 72.15+0.81% and 60.57+4.27% at 48-hours, 57.63+4.54% and 60.65+4.90% at 72-hours.
Conclusion: MSC-CM appears to inhibit fibroblast to myofibroblast differentiation, over-riding the pro-differentiation effects of Wnt-3a and TGF-β1. Whilst both TGF-β1 and Wnt-3a have emerged as key players in IPF pathogenesis, we are the first to demonstrate that MSC-CM may be crucial in modulating their pro-fibrogenic effects. These actions of MSC-CM demonstrate exploitative potential for future anti-IPF therapeutic strategies.
Keywords: Mesenchymal stem cells; Idiopathic pulmonary fibrosis; Wnt- 3a; TGF-1; Fibrosis; Mesenchymal stem cell conditioned media; Alpha smooth muscle actin; Myofibroblast differentiation
Introduction
Background
Idiopathic pulmonary fibrosis is a chronic progressive incurable fibrotic disease with a very poor median life expectancy of 3-5 years [1-3]. The current hypothesis of the pathophysiology of IPF dictates that the mechanisms responsible comprise of repeated alveolar epithelial injury and an aberrant fibrotic process with fibroblast to myofibroblast differentiation and formation of the fibroblastic foci playing a significant role in the development of the IPF. It is postulated that the aberrant wound repair and fibrotic processes are regulated by a plethora of growth factors and cytokines [1,2,4-6].
Several new growth factors have been implicated as having a potentially significant influence on IPF pathogenesis. TGF-β1 has been documented to have key pro-fibrotic properties and influence fibroblast to myofibroblast differentiation [7-10]. Alongside TGF-β1, more recently the Wnt family of growth factors has been postulated to also influence fibrosis and IPF [11-14]. The secreted glycoprotein Wnt is recognised in the literature for its substantial influence in the development of colorectal cancer and also during embryogenesis and organogenesis [15-17]. In addition to these well-known functions, Wnt-3a, other members of the Wnt family and TGF-β1 have also been shown to effect several aspects of IPF pathogenesis including fibroblast to myofibroblast differentiation and the Epithelial To Mesenchymal Transition (EMT) [11,13]. Furthermore, in vitro studies have now determined that Wnt-3a can induce murine fibroblast to myofibroblast differentiation as confirmed through demonstrating the presence of α-SMA expression [18,19]. Therefore if this effect of Wnt-3a on fibroblast to myofibroblast differentiation could be replicated in human IPF lung fibroblasts, this would provide further evidence that Wnt-3a may have a significant involvement in the key process of myofibroblast differentiation, thereby playing a possible role in the formation of the fibroblastic foci in IPF. Thus if the actions of Wnt-3a and TGF-β1 could be suppressed then this may provide an avenue at which future therapeutic agents could be targeted.
Mesenchymal stem cells are multipotent stem cells capable of differentiation into cells of the chondrogenic, adipogenic and osteogenic lineages and have been studied as a potential therapeutic agent for the treatment of IPF and fibrotic diseases [20-23]. In addition to their differentiation potential, MSC also secrete a plethora of growth factors and cytokines which have been shown to demonstrate paracrine mediated anti-inflammatory and anti-fibrotic effects in vitro and in vivo [24-28]. Therefore MSC-CM, which contains the secretory proteins of the MSC, may have a therapeutic role in the treatment of IPF.
Current evidence has demonstrated that TGF-β1 has a significant influence in the development of IPF; in addition studies have also demonstrated that Wnt-3a has a potential involvement in IPF pathogenesis and may regulate fibroblast to myofibroblast differentiation. Furthermore, MSC have been shown to attenuate fibrosis possibly through paracrine mechanisms; however the exact mechanism as to how MSC achieve this anti-fibrotic effect is as yet unclear and the effect of MSC-CM on Wnt-3a and TGF-β1 induced fibrosis has yet to be explored.
Through our in vitro fibroblast differentiation model, we first induced human lung fibroblast to myofibroblast differentiation with Wnt-3a and TGF-β1, then we ascertained that MSC-CM could inhibit this key differentiation process and additionally we also determined that MSC-CM could de-differentiate TGF-β1 induced myofibroblasts back to fibroblasts. The results of this study provide further insight into the possible paracrine mediated anti-fibrotic effects of MSC and further support the possible use of MSC-CM as a potential therapeutic agent in the treatment of IPF.
Method
Fibroblast culture
Normal human lung fibroblast (CCD-8Lu) and IPF lung fibroblast (LL29) were purchased from ATCC, Rockville USA, maintained in continuous culture with DMEM supplemented with 10% FBS, 1% L-glutamine and 1% Non-Essential Amino Acid (NEAA) (Lonza, Belgium) and utilised between passages 7-14 and passages 8-16 respectively for all experiments.
MSC isolation, culture and conditioned media preparation
MSCs were isolated from human bone-marrow, propagated and characterized following previously described methodology [28] with little modification. Briefly, human whole bone marrow aspirate (collected from iliac crest) (Lonza, USA) was seeded at a density of 105 mononuclear cells/cm2 on 10 ng/ml Fibronectin-coated (Cat. No. F0895, Sigma) T-75 tissue culture flasks in 20 ml of DMEM supplemented with 5% FBS, 1% L-glutamine, 1% NEAA and 1% PSA (Penicillin-Streptomycin- Amphotericin B). Cells were maintained in continuous culture for three weeks in a humidified incubator at 37�C in the presence of 5% CO2 and 95% air. After 7 days, media was replaced with antibiotic-free medium as described above. Media was replenished after a further week. At the end of third week, the adherent hMSC population was harvested with trypsin and passaged subsequently using 1:4 split ratios for expansion. Isolated human MSCs (passage 1) were placed on 24-well plates and grown to 80- 90% confluency for Immunophenotyping. Cells were fixed with 4% paraformaldehyde, blocked with 3% BSA (Bovine serum albumin), and characterised using the human MSC characterisation kit containing anti-human mouse anti-CD44, anti-CD90, anti-CD146, anti-CD14, anti-CD19 and anti-STRO-1 primary antibodies at 1:500 dilution (Cat. No. SCR067, Millipore). Cells were incubated with primary antibodies at 4�C overnight. Secondary antibodies were antimouse IgG-NL557 for all except STRO-1 and anti-mouse IgM-NL493 for STRO-1 (both at 1:200 dilutions) (R & D System). For functional characterisation, isolated MSCs were differentiated into osteogenesis, adipogenesis and chondrogenesis lineages using StemPro® hMSC differentiation kits following the manufacturer�s instructions (Gibco, Invitrogen, USA) as per previously described methodology [28]. Osteogenesis, adipogenesis and chondrogenesis were confirmed by traditional validated cytological staining with Alizarin Red, Oil Red O and Alcian Blue as well as by immunostaining with anti-osteocalcin, anti-FABP-4 and anti-aggrecan antibodies respectively following manufacturer�s protocol (Human MSC Functional Identification Kit, Cat. No- SC006, R & D System). For visualisation, secondary antibodies were used; anti-mouse IgG-NL557 for osteocalcin and anti-goat IgG-NL493 for FABP-4 and aggrecan (both at 1:200 dilutions) (NorthernLights, R & D System). DAPI was used for nuclear staining in all immunocytochemistry assays. Images were acquired by a fluorescent microscope (Nikon Eclipse Ti-ST, Japan). MSC-CM was prepared following previously described methodology [28]. Briefly, MSCs (passage 1�3) were grown to 80-90% confluency in T75 tissue culture flasks using 5% FBS supplemented complete DMEM media. Cells were washed once with PBS and twice with serum-free DMEM (SF-DMEM) to remove any serum. MSCs were conditioned by exposing SF-DMEM on 80-90% confluent MSC for 24 hours in standard culture condition. SF-MSC CM was collected, centrifuged to remove any cell debris and stored at -80�C and sterile filtered prior to use. Through immunophenotyping the cells were confirmed to be 100% MSC.
TGF-β1 and Wnt-3a induced fibroblast to myofibroblast differentiation
The in vitro differentiation of fibroblast to myofibroblast was achieved using treatment with TGF-β1 and Wnt-3a. Initially CCD- 8Lu and LL29 cells were cultured in complete growth media on a 48- well plate for 24 hours then incubated for a further 24 hours in SFDMEM. Following this, the cells were treated with 600�l of 5ng/ml of TGF-β1 in SF-DMEM for 24 hours and 600�l of 300ng/ml of Wnt-3a in SF-DMEM for 72 hours. To detect myofibroblast differentiation following treatment with TGF-β1 and Wnt-3a, immunocytochemical analysis to detect the presence of the myofibroblast marker α-SMA was performed using the FITC-conjugated α-SMA antibody stain (Sigma Aldrich UK) at a concentration of 1:500. DAPI at a concentration of 1:500 was utilised for nuclear staining and TRITCconjugated phalloidin antibody stain at a concentration of 1:500 was utilised to detect F-Actin. Subsequent images were captured using an Eclipse Ti-S immunofluorescence microscope (Nikon® Japan) and, NIS elements 3.2 (Nikon Japan) image capture software. The images were then merged using Photoshop CS4 (Adobe® USA) image editing software to allow image analysis. Subsequent cell counting was performed on the images to determine the percentage of fibroblasts that had differentiated into myofibroblasts.
Co-administration of MSC-CM and TGF-β1 on fibroblasts
To evaluate the effect MSC-CM has on the TGF-β1 induced fibroblast to myofibroblast differentiation process, co-administration of MSC-CM with TGF-β1 was performed on CCD-8Lu and LL29 fibroblasts. Administration of TGF-β1 alone and administration of SF-DMEM alone were utilised as the positive and negative controls respectively. The fibroblasts were cultured in a 48-well plate for 24 hours; the cells were then incubated in SF-DMEM for a further 24 hours. TGF-β1 was diluted in MSC-CM to a concentration of 5ng/ ml. TGF-β1 was also diluted in SF-DMEM to a concentration of 5ng/ ml. 600�l of TGF-β1 in MSC-CM per well, 600�l of TGF-β1 in SFDMEM per well and 600�l of SF-DMEM alone per well were added to the fibroblasts. Following addition of the solutions, the cells were returned to the incubator for 24 hours to allow cell differentiation to occur. Upon completion of the 24 hours, immunocytochemical analysis was performed and the percentage of differentiated myofibroblasts quantified as described previously.
Co-administration of MSC-CM and Wnt-3a on fibroblasts
To assess the effect that MSC-CM has on the Wnt-3a- induced fibroblast to myofibroblast differentiation process, Wnt-3a and MSCCM were co-administered on CCD-8Lu and LL29 fibroblasts for 72 hours. Treatment with Wnt-3a alone and treatment with SF-DMEM were utilised as the positive and negative controls respectively. The fibroblasts were cultured in a 48-well plate for 24 hours then incubated in SF-DMEM for a further 24 hours. Following this 600�l of 300ng/ ml Wnt-3a with MSC-CM were added, 600�l of 300ng/ml Wnt-3a in SF-DMEM and 600�l of SF-DMEM were also added to the wells. The plates were incubated for a further 72 hours to allow fibroblast to myofibroblast differentiation to occur. Upon completion of the 24 hours, immunocytochemical analysis was performed and the percentage of differentiated myofibroblasts quantified as described previously.
Administration of MSC-CM to TGF-β1 differentiated myofibroblasts
Following the exploration of the effects of MSC-CM on the fibroblast to myofibroblast differentiation process, the next step was to determine the effect that administration of MSC-CM has on myofibroblasts. To evaluate this, SF-DMEM alone were added to the undifferentiated fibroblasts to act as a negative control, the CCD-8Lu and LL29 fibroblasts were subsequently differentiated into myofibroblasts with 600�l of 5ng/ml of TGF-β1. Following this, immunofluorescence analysis was conducted to detect the presence of α-SMA expression and thus confirm that the cells had differentiated into myofibroblasts. This stage where the cells were now confirmed to be myofibroblasts was designated as baseline. At baseline, 500�l of MSC-CM per well were added to the myofibroblasts. 500�l of SFDMEM per well were also added to the myofibroblasts to determine if there was any natural de-differentiation of the myofibroblasts and thus acted as a positive control. Twenty-four, 48 and 72 hours after administration of MSC-CM, the plates were fixed and stained with the immunofluorescence antibody stains and the number of cells expressing α-SMA was quantified as described previously.
Results
MSC-CM inhibits TGF-β1 induced fibroblast to myofibroblast differentiation
First, MSC-CM was prepared using isolated human bone marrow MSC which were immunophenotypically CD44+, CD90+, CD146+ , STRO-1+, CD14- , and CD19- (Figure 1) and underwent differentiation into osteogenesis, adipogenesis and chondrogenesis lineages (Figure 1). To evaluate the effect of MSC-CM on the TGF-β1 induced CCD-8Lu and LL29 fibroblast to myofibroblast differentiation process; MSC-CM was co-administered with TGF-β1 on cultured CCD-8Lu and LL29 cells for 24 hours. These results demonstrate that 24 hours treatment with TGF-β1 resulted in significant (p<0.001) fibroblast to myofibroblast differentiation as confirmed through the presence of α-SMA expression compared to control (SF-DMEM) in both CCD-8Lu and LL29 cells (Figure 2). However the results clearly demonstrate that 24 hours coadministration of MSC-CM with TGF-β1 resulted in a significant reduction (p<0.001) in the percentage of CCD-8Lu cells expressing α-SMA in comparison to TGF-β1 treatment alone. The addition of MSC-CM resulted in a 31.40±1.44% reduction when compared with the TGF-β1 only treated sample (MSC-CM+TGF-β1 54.2±1.8%, TGF-β1 78.90±2.80%) (Figure 2 A/C). The results for the LL29 cells are reflective of the CCD-8Lu cells and illustrate that 24 hours coadministration of MSC-CM with TGF-β1 resulted in a significant reduction (p<0.001) in the percentage of cells expressing α-SMA in comparison to TGF-β1 treatment alone (Figure 2). The addition of MSC-CM resulted in a 35.69±7.47% reduction when compared with the TGF-β1 only treated sample (MSC-CM + TGF-β1 51.8±3.0%, TGF-β1 80.90±5.60%) (Figure 2 A/B). Therefore, the results conclude that MSC-CM abrogates TGF-β1 induced fibroblast to myofibroblast differentiation. Furthermore, this suppressive effect of MSC-CM is more pronounced on LL29 cells than CCD-8Lu cells by 1.13-fold.

Figure 1: Immunophenotyping of MSCs isolated from human bone marrow
aspirates display positive staining for mesenchymal stem cell markers
CD44, STRO-1, CD90 and CD146 (A,B,C,D); and negative staining
for haematopoietic markers CD14 and CD19 (E,F). MSC tri-lineage
differentiation: osteogenesis (G,J), adipogenesis (H,K) and chondrogenesis
(I,L) were confirmed by cytochemical/immunocytochemistry staining with
Alizarin Red/anti-Osteocalcin, Oil Red O/anti-FABP-4 and Alcian Blue/anti-
Aggrecan respectively. Cells were confirmed to be 100% MSC. Scale bars,
100 �m.
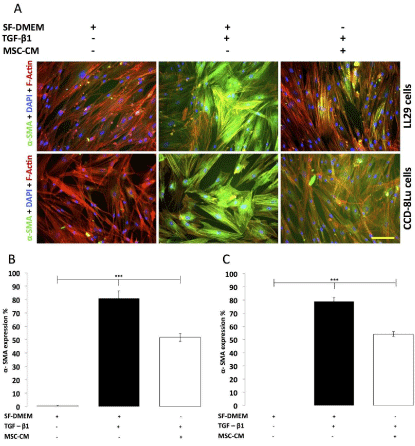
Figure 2: The effect of MSC-CM on TGF-β1 induced fibroblast to myofibroblast
differentiation (CCD-8Lu and LL29). (A) Immunofluorescence images of
fibroblasts following 24 hours treatment with MSC-CM and TGF-β1. Scale
Bar = 100�m. (B) Percentage of LL29 cells expressing α-SMA after 24 hours
treatment. Percentage of cells expressing α-SMA after treatment with SFDMEM
in the absence of TGF-β1 (grey bar), percentage of cells expressing
α-SMA after treatment with TGF-β1 5ng/ml (black bar), percentage of cells
expressing α-SMA following co-administration of MSC-CM and TGF-β1
(white bar) (n=3 per group, ***p<0.001). (C) Percentage of CCD-8Lu cells
expressing α-SMA after 24 hours treatment. Percentage of cells expressing
α-SMA after treatment with SF-DMEM in the absence of TGF-β1 (grey bar),
percentage of cells expressing α-SMA after treatment with TGF-β1 5ng/ml
(black bar), percentage of cells expressing α-SMA following co-administration
of MSC-CM and TGF-β1 5ng/ml (white bar) (n=3 per group, ***p<0.001).
MSC-CM inhibits Wnt-3a induced fibroblast to myofibroblast differentiation
To evaluate the effect of MSC-CM on the Wnt-3a-induced CCD- 8Lu and LL29 fibroblast to myofibroblast differentiation process; MSC-CM was co-administered with Wnt-3a for 72 hours. The results illustrate that 72 hours incubation with Wnt-3a resulted in significant (p<0.001) fibroblast to myofibroblast differentiation as confirmed through the presence of α-SMA expression in comparison to control (SF-DMEM) in both CCD-8Lu and LL29 cells (Figure 3). However, the results clearly demonstrate that co-administration of Wnt-3a and MSC-CM for 72 hours resulted in a significant reduction (p<0.001) in the percentage of CCD-8Lu cells expressing α-SMA in comparison to Wnt-3a treatment alone. The addition of MSC-CM to Wnt-3a resulted in an 80.80±3.60% reduction when compared to the Wnt- 3a only treated sample (MSC-CM+Wnt-3a 10.20±1.90%, Wnt-3a 52.90±0.20%) (Figure 3A/C). As observed with TGF-β1, the results for the LL29 cells are reflective of the CCD-8Lu cells. The results illustrate that co-administration of Wnt-3a and MSC-CM to LL29 cells for 72 hours resulted in a significant reduction (p<0.001) in the percentage of cells expressing α-SMA in comparison to Wnt-3a treatment alone. The addition of MSC-CM to Wnt-3a resulted in a 79.70±3.90% reduction when compared to the Wnt-3a only treated sample (MSCCM+ Wnt-3a 11.30±2.20%, Wnt-3a 55.60±5.90%) (Figure 3A/B). Therefore, MSC-CM has a significant suppressive effect on the Wnt-3a induced fibroblast to myofibroblast differentiation process. Furthermore, this suppressive effect of MSC-CM appears to be very similar between CCD-8Lu and LL29 cells.

Figure 3: The effect of MSC-CM on Wnt-3a induced fibroblast to myofibroblast
differentiation (CCD-8Lu and LL29). (A) Immunofluorescence images of
fibroblasts following 72 hours treatment with MSC-CM and Wnt-3a. Scale
Bar = 100�m. (B) Percentage of LL29 cells expressing α-SMA after 72 hours
treatment. Percentage of cells expressing α-SMA after treatment with SFDMEM
in the absence of Wnt-3a (grey bar), percentage of cells expressing
α-SMA after treatment with Wnt-3a 300ng/ml (black bar), percentage of
cells expressing α-SMA following co-administration of MSC-CM and Wnt-3a
(white bar) (n=3 per group, ***p<0.001). (C) Percentage of CCD-8Lu cells
expressing α-SMA after 72 hours treatment. Percentage of cells expressing
α-SMA after treatment with SF-DMEM in the absence of Wnt-3a (grey bar),
percentage of cells expressing α-SMA after treatment with Wnt-3a 300ng/ml
(black bar), percentage of cells expressing α-SMA following co-administration
of MSC-CM and Wnt-3a 300ng/ml (white bar) (n=3 per group, ***p<0.001.
MSC-CM de-differentiates CCD-8Lu and LL29 derived myofibroblasts
CCD-8Lu and LL29 fibroblasts were differentiated into myofibroblasts with 24 hours TGF-β1 treatment; this stage where the cells were now myofibroblasts was designated as baseline. Fibroblasts were also treated in parallel with SF-DMEM alone as a negative control and showed negligible α-SMA expression throughout (Figure 4). Prior to MSC-CM administration at baseline, there was no significant difference in the percentage of CCD-8Lu cells expressing α-SMA between the MSC-CM treated and control group (SF-DMEM) (Figure 4C/D). Furthermore, MSC-CM did not show any effect on the percentage of cells expressing α-SMA after 24 hours treatment in comparison to control (SF-DMEM) (Figure 4C/D). As observed previously the LL29 cells follow a similar pattern to that of the CCD-8Lu cells. Prior to MSC-CM administration at baseline, there was no significant difference in the percentage of LL29 cells expressing α-SMA between the MSC-CM treated and their corresponding control group (SF-DMEM) (Figure 4A/B). However 24, 48 and 72 hours treatment with MSC-CM did show a significant reduction (p<0.001) in the percentage of cells expressing α-SMA which were 15.10±5.60%, 25.60±3.30%, 24.70±8.10% respectively, in comparison to their corresponding control (SF-DMEM) (MSC-CM 24 hours 73.30±0.70%, 48 hours 60.70±3.00%, 72 hours 60.40±3.50%, SF-DMEM 24 hours 86.50±5.00%, 48 hours 81.50±1.20%, 72 hours 80.60±4.10%) (Figure 4A/B). The observations from this experiment suggest that MSC-CM de-differentiates myofibroblasts back to fibroblasts in a duration exposure-dependent manner. Furthermore, it appears that the effect of MSC-CM on the de-differentiation of myofibroblasts occurs earlier within LL29 at 24 hours, in comparison to CCD-8Lu at 48 hours after MSC-CM administration.
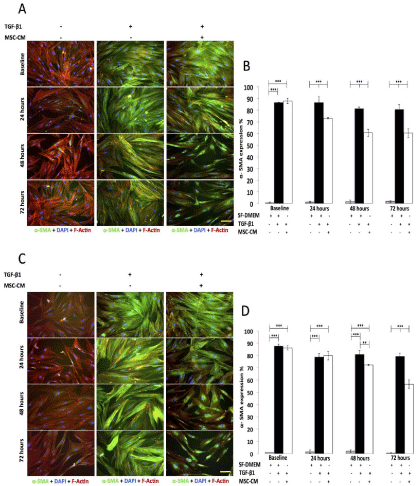
Figure 4: The effect of MSC-CM on myofibroblasts (CCD-8Lu and LL29).
(A) Immunofluorescence images of myofibroblasts (LL29) after 72 hours
treatment with MSC-CM. (B) Percentage of myofibroblasts (LL29) expressing
α-SMA after 72 treatments with MSC-CM. Percentage of cells expressing
α-SMA with SF-DMEM in the absence of TGF-β1 and MSC-CM (grey bars).
Percentage of cells expressing α-SMA follows TGF-β1 treatment alone (black
bars). Percentage of cells expressing α-SMA follows treatment with MSCCM
(white bars). (n=3 per group, ***p-<0.001). (C) Immunofluorescence
images of myofibroblasts (CCD-8Lu) after 72 hours treatment with MSCCM.
(D) Percentage of myofibroblasts (CCD-8Lu) expressing α-SMA after
72 treatments with MSC-CM. Percentage of cells expressing α-SMA with
SF-DMEM in the absence of TGF-β1 and MSC-CM (grey bars). Percentage
of cells expressing α-SMA follows TGF-β1 treatment alone (black bars).
Percentage of cells expressing α-SMA follows treatment with MSC-CM (white
bars). (n=3 per group, **p<0.01, ***p-<0.001).
Discussion
Idiopathic pulmonary fibrosis is a complex disease about which there is much we still do not yet understand. However through continuing research, we have developed our knowledge of IPF pathogenesis and have now ascertained that the fibroblastic foci are a focal point of the fibrogenesis in IPF [1,5,29,30]. The major cell population within the fibroblastic foci are the myofibroblasts; thus a key contributor to the development of the fibroblastic foci is fibroblast to myofibroblast differentiation with a resultant increase in the myofibroblast population [1,30-32]. Consequently any explorative approach attempting to suppress or limit this fibroblast to myofibroblast differentiation may potentially help to reduce the activated myofibroblast population and attenuate the active profibrotic processes from occurring in IPF. It is currently postulated that in IPF, myofibroblast differentiation is under the regulation of growth factors, the best documented of which is TGF-β [31-34]. However recent evidence has also suggested that this process may be under the influence of further growth factors, the most prominent of which is Wnt-3a [13,18,19,35]. Further evidence of the Wnt signalling pathway having a pro-fibrotic influence has been documented in cardiac [36], hepatic [37] and renal [38] fibrosis.
Presently there are no effective pharmaceutical therapies effective at halting the fibrotic process in IPF. Due to this lack of therapeutic options, research into a regenerative therapeutic approach utilising MSC has been strongly considered as a potential treatment modality [20,39]. Mesenchymal stem cells have been demonstrated to ameliorate fibrosis within renal, cardiac and hepatic animal models of fibrosis [36,40-45]. Additional evidence that MSC may possess atherapeutic role in IPF has been presented through the conduction of in vitro studies, which have demonstrated that MSC and MSC-CM can attenuate fibrosis within animal models of IPF [21-23,46,47]. In addition to the current literature, our results have provided further evidence that MSC-CM has anti-fibrotic effects with the ability to modulate the fibroblast to myofibroblast differentiation process.
We have demonstrated for the first time that Wnt-3a can induce fibroblast to myofibroblast differentiation in adult human normal and IPF lung fibroblasts; thus providing further evidence for the potential involvement of Wnt-3a in IPF. More importantly, we have determined for the first time that MSC-CM significantly suppresses the Wnt-3a-induced fibroblast to myofibroblast differentiation process in human and IPF lung fibroblasts. However, the mechanism of action as to how MSC-CM exerted its effects on Wnt-3a-induced myofibroblast differentiation requires further exploration. Wnt-3a is a secreted glycoprotein that binds to frizzled receptors resulting in the intracellular release of cytoplasmic β-catenin from the GSK3β complex. β-catenin then migrates to the nucleus, binds to its nuclear LEF/TCF receptor and exerts its biological effect [13,14,35]. Therefore, due to the complex mechanism of Wnt signalling, there may be several molecules in the Wnt signalling pathway which could be prone to inhibitory modulation. Studies have determined that FGF can inhibit the canonical Wnt signalling pathway [48] through inhibiting the formation of the -catenin LEF/TCF complex. Furthermore, upregulating Dickopf-1 an inhibitor of Wnt-3a signalling has not only been shown to inhibit but also reverse bleomycin induced lung fibrosis [13]. Although these above studies did not utilise MSC or MSC-CM specifically, they all demonstrate that not only is the inhibition of Wnt signalling possible, but also that inhibition of this signalling pathway can inhibit cell differentiation. Our work has not only provided further support for the involvement of Wnt-3a in IPF but also demonstrated that MSC-CM has the ability to inhibit Wnt-3a induced differentiation, possibly through the mechanisms discussed above.
In accordance with current literature [8-10] our work has demonstrated that TGF-β1 can induce human normal and IPF lung fibroblast to myofibroblast differentiation. Additionally, for the first time we have demonstrated that MSC-CM can inhibit this TGF-β1 induced differentiation process and also de-differentiate myofibroblasts. However the question remains as to the mechanism of action that is responsible for this observed MSC-CM suppression of myofibroblastic differentiation and de-differentiation. Although this study is the first to demonstrate that MSC-CM can supress TGF-β1-induced myofibroblast differentiation, several previous studies have aimed to isolate and inhibit specific components of the TGF-β1 signalling pathway in an attempt to disrupt its profibrotic actions with some promising results. Prostaglandin E2 [49], interleukin 7 [50], FGF [51,52] have been demonstrated to inhibit TGF-β1 and subsequent fibroblast differentiation through Smad dependent and independent pathways. Furthermore following fibroblast to myofibroblast differentiation, it was postulated that the myofibroblast was a terminally differentiated cell and in the process of normal wound repair, a reduction in the myofibroblast population occurred through myofibroblast apoptosis [1,19,53,54]. This theory has been challenged and it is now postulated that myofibroblast dedifferentiation may occur. PDGF [55] and FGF-1 and FGF-2 have been shown to cause de-differentiation of myofibroblasts [56]. The results of our study further support the de-differentiation potential of myofibroblasts. Furthermore MSC have been shown to secrete both FGF and PGE and PDGF [6]. Therefore it is possible that these growth factors were evident within our MSC-CM and responsible for the suppressive effects and de-differentiate effects that were observed.
Conclusion
Fibroblast to myofibroblast differentiation is believed to be a key mechanism in the pathogenesis of IPF. Although MSC-CM has been shown to exert anti-inflammatory and anti-fibrotic effects, the underlying mechanism of action has yet to be fully elucidated. Our data provides further evidence for the involvement of TGF-β1 and Wnt-3a in IPF and for the first time provides a potential mechanism of action through which MSC-CM may exert its anti-fibrotic effects.
References
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011; 378: 1949-1961.
- Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res. 2002; 3.
- Selman M, King TE, Pardo A. American Thoracic Society, European Respiratory Society, American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001; 134: 136-151.
- Pardo A, Selman M. Idiopathic pulmonary fibrosis: new insights in its pathogenesis. Int J Biochem Cell Biol. 2002; 34: 1534-1538.
- Zoz DF, Lawson WE, Blackwell TS. Idiopathic pulmonary fibrosis: a disorder of epithelial cell dysfunction. Am J Med Sci. 2011; 341: 435-438.
- Hass R, Kasper C, Bohm S, Jacobs R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal. 2011; 9: 12.
- Popova AP, Bozyk PD, Goldsmith AM, Linn MJ, Lei J, Bentley JK, et al. Autocrine production of TGF-beta1 promotes myofibroblastic differentiation of neonatal lung mesenchymal stem cells. Am J Physiol Lung Cell Mol Physiol. 2010; 298: 735-743.
- Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004; 18: 816-827.
- Evans RA, Tian YC, Steadman R, Phillips AO. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res. 2003; 282: 90-100.
- Watts KL, Sampson EM, Schultz GS, Spiteri MA. Simvastatin inhibits growth factor expression and modulates profibrogenic markers in lung fibroblasts. Am J Respir Cell Mol Biol. 2005; 32: 290-300.
- Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012; 3: 735.
- Emblom-Callahan MC, Chhina MK, Shlobin OA, Ahmad S, Reese ES, Iyer EP, et al. Genomic phenotype of non-cultured pulmonary fibroblasts in idiopathic pulmonary fibrosis. Genomics. 2010; 96: 134-145.
- Henderson WR Jr, Chi EY, Ye X, Nguyen C, Tien YT, Zhou B, et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A. 2010; 107: 14309-14314.
- Shafer SL, Towler DA. Transcriptional regulation of SM22alpha by Wnt3a: convergence with TGFbeta(1)/Smad signaling at a novel regulatory element. J Mol Cell Cardiol. 2009; 46: 621-635.
- Clevers H. Wnt breakers in colon cancer. Cancer Cell. 2004; 5: 5-6.
- Myant K, Sansom OJ. Wnt/Myc interactions in intestinal cancer: partners in crime. Exp Cell Res. 2011; 317: 2725-2731.
- Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003; 1653: 1-24.
- Carthy JM, Garmaroudi FS, Luo Z, McManus BM. Wnt3a induces myofibroblast differentiation by upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PLoS One. 2011; 6: 19809.
- Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007; 132: 1311-1321.
- Abreu SC, Antunes MA, Pelosi P, Morales MM, Rocco PR. Mechanisms of cellular therapy in respiratory diseases. Intensive Care Med. 2011; 37: 1421-1431.
- Antoniou KM, Papadaki HA, Soufla G, Kastrinaki MC, Damianaki A, Koutala H, et al. Investigation of bone marrow mesenchymal stem cells (BM MSCs) involvement in Idiopathic Pulmonary Fibrosis (IPF). Respir Med. 2010; 104: 1535-1542.
- Kumamoto M, Nishiwaki T, Matsuo N, Kimura H, Matsushima K. Minimally cultured bone marrow mesenchymal stem cells ameliorate fibrotic lung injury. Eur Respir J. 2009; 34: 740-748.
- Moodley Y, Atienza D, Manuelpillai U, Samuel CS, Tchongue J, Ilancheran S, et al. Human umbilical cord mesenchymal stem cells reduce fibrosis of bleomycin-induced lung injury. Am J Pathol. 2009; 175: 303-313.
- Baksh D, Yao R, Tuan RS. Comparison of proliferative and multilineage differentiation potential of human mesenchymal stem cells derived from umbilical cord and bone marrow. Stem Cells. 2007; 25: 1384-1392.
- Baksh D, Song L, Tuan RS. Adult mesenchymal stem cells: characterization, differentiation, and application in cell and gene therapy. J Cell Mol Med. 2004; 8: 301-316.
- Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008; 2: 313-319.
- Akram KM1, Samad S, Spiteri M, Forsyth NR . Mesenchymal stem cell therapy and lung diseases. Adv Biochem Eng Biotechnol. 2013; 130: 105-129.
- Akram KM, Samad S, Spiteri MA, Forsyth NR. Mesenchymal stem cells promote alveolar epithelial cell wound repair in vitro through distinct migratory and paracrine mechanisms. Respir Res. 2013; 14: 9.
- Aggarwal D, Mohapatra PR. Dyspnoea: a prognostic marker for idiopathic pulmonary fibrosis. Eur Respir J. 2011; 37: 476.
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011; 183: 788-824.
- Caminati A, Harari S. IPF: New insight in diagnosis and prognosis. Respir Med. 2010; 104: 2-10.
- Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2010; 4: 367-388.
- Fernandez Perez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 2010; 137: 129-137.
- Harari S, Caminati A. IPF: new insight on pathogenesis and treatment. Allergy. 2010; 65: 537-553.
- K�nigshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, et al. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS One. 2008; 3: 2142.
- Chen Q, Harding SE, Ali NN, Lyon AR, Boccaccini AR. Biomaterials in cardiac tissue engineering: Ten years of research survey. Materials Science and Engineering: R: Reports 2008; 59: 1-37.
- Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K, et al. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008; 294: 39-49.
- Surendran K, McCaul SP, Simon TC. A role for Wnt-4 in renal fibrosis. Am J Physiol Renal Physiol. 2002; 282: 431-441.
- Burdon TJ, Paul A, Noiseux N, Prakash S, Shum-Tim D. Bone marrow stem cell derived paracrine factors for regenerative medicine: current perspectives and therapeutic potential. Bone Marrow Res. 2011; 2011: 207326.
- Semedo P, Correa-Costa M, Antonio Cenedeze M, Maria Avancini CM, Antonia dR, Shimizu MH, et al. Mesenchymal stem cells attenuate renal fibrosis through immune modulation and remodeling properties in a rat remnant kidney model. Stem Cells. 2009; 27: 3063-3073.
- Morigi M, Imberti B, Zoja C, Corna D, Tomasoni S, Abbate M, et al. Mesenchymal stem cells are renotropic, helping to repair the kidney and improve function in acute renal failure. J Am Soc Nephrol. 2004; 15: 1794-1804.
- Gandolfi F, Vanelli A, Pennarossa G, Rahaman M, Acocella F, Brevini TA. Large animal models for cardiac stem cell therapies. Theriogenology. 2011; 75: 1416-1425.
- Christoforou N, Gearhart JD. Stem cells and their potential in cell-based cardiac therapies. Prog Cardiovasc Dis. 2007; 49: 396-413.
- Sato Y, Araki H, Kato J, Nakamura K, Kawano Y, Kobune M, et al. Human mesenchymal stem cells xenografted directly to rat liver are differentiated into human hepatocytes without fusion. Blood. 2005; 106: 756-763.
- Aquino JB, Bolontrade MF, Garc�a MG, Podhajcer OL, Mazzolini G. Mesenchymal stem cells as therapeutic tools and gene carriers in liver fibrosis and hepatocellular carcinoma. Gene Ther. 2010; 17: 692-708.
- Tzouvelekis A, Koliakos G, Ntolios P, Baira I, Bouros E, Oikonomou A, et al. Stem cell therapy for idiopathic pulmonary fibrosis: a protocol proposal. J Transl Med. 2011; 9: 182.
- Cargnoni A, Ressel L, Rossi D, Poli A, Arienti D, Lombardi G, et al. Conditioned medium from amniotic mesenchymal tissue cells reduces progression of bleomycin-induced lung fibrosis. Cytotherapy. 2012; 14: 153-161.
- Ambrosetti D, Holmes G, Mansukhani A, Basilico C. Fibroblast growth factor signaling uses multiple mechanisms to inhibit Wnt-induced transcription in osteoblasts. Mol Cell Biol. 2008; 28: 4759-4771.
- Thomas PE, Peters-Golden M, White ES, Thannickal VJ, Moore BB. PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am J Physiol Lung Cell Mol Physiol. 2007; 293: L417-428.
- Huang M, Sharma S, Zhu LX, Keane MP, Luo J, Zhang L, et al. IL-7 inhibits fibroblast TGF-beta production and signaling in pulmonary fibrosis. J Clin Invest. 2002; 109: 931-937.
- Greenberg RS, Bernstein AM, Benezra M, Gelman IH, Taliana L, Masur SK. FAK-dependent regulation of myofibroblast differentiation. FASEB J. 2006; 20: 1006-1008.
- Cushing MC, Mariner PD, Liao JT, Sims EA, Anseth KS. Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. FASEB J. 2008; 22: 1769-1777.
- Singh SR, Hall IP. Airway myofibroblasts and their relationship with airway myocytes and fibroblasts. Proc Am Thorac Soc. 2008; 5: 127-132.
- Gu L, Zhu YJ, Yang X, Guo ZJ, Xu WB, Tian XL. Effect of TGF-beta/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol Sin. 2007; 28: 382-391.
- Hecker L, Jagirdar R, Jin T, Thannickal VJ. Reversible differentiation of myofibroblasts by MyoD. Exp Cell Res. 2011; 317: 1914-1921.
- Maltseva O, Folger P, Zekaria D, Petridou S, Masur SK. Fibroblast growth factor reversal of the corneal myofibroblast phenotype. Invest Ophthalmol Vis Sci. 2001; 42: 2490-2495.