
Research Article
J Stem Cells Res, Rev & Rep. 2015;2(1): 1018.
Co-Localization of CD133/EGFR and Antiangiogenic Activity Drive the Antitumor Effect of Nimotuzumab and Radiation in Human GBM U87MG Xenografted in Mice
Diaz-Miqueli A¹*, Markelova MR², Lemm M² and Fichtner I²
¹Department of System Biology, Center of Molecular Immunology, Cuba
²Department of Experimental Pharmacology, Max Delbrück Center for Molecular Medicine, Germany
*Corresponding author: Diaz-Miqueli A, Department of System Biology, Center of Molecular Immunology, 216 St. Havana, Cuba, Tel: 537-271 5057; Fax: 537-273 3509; Email: arlhee@cim.sld.cu
Received: December 12, 2014; Accepted: February 23, 2015; Published: February 25, 2015
Abstract
The targeting of CD133 radio and chemoresistant population have been suggested a crucial strategy to improve the local control disease in the treatment of brain tumors. In this study nimotuzumab, a monoclonal antibody specific to EGFR, showed no significant cytotoxic activity in vitro, neither alone nor in combination with radiotherapy against the human glioma cells U87MG assessed by MTT assays. In contrast, the co-administration of nimotuzumab and radiation significantly delayed subcutaneous tumor growth in NMRI nude mice, together with a significant antiangiogenic response. In vitro, U87MG cells formed tumor neurospheres when cultured in serum-free neural stem cell medium. Moreover, tumor neurospheres, but not adherent cells, showed an increased immunoreactivity to the neural CSC marker CD133. Immunofluorescence analysis performed to examine protein expression showed that CD133 was co-localized with the EGFR, suggesting a potential molecular mechanism by which nimotuzumab is able to target the CD133 radioresistant population in the U87MG cell line. In summary, the present study suggests that antitumor activity of nimotuzumab in combination with radiation against U87MG xenografts is mediated, at least in part, by a potent antiangiogenic response in addition to the ability of nimotuzumab to target the CD133 radioresistant population, suggesting a promising alternative to the failure of current available therapies in the treatment of glioblastoma.
Keywords: CD133; Epidermal growth factor receptor; Glioblastoma multiform; Nimotuzumab; Radiation; U87MG
Abbreviations
CSC: Cancer Stem Cells; EGF: Epidermal Growth Factor; EGFR: EGF Receptor; GBM: Glioblastoma Multiform; MGMT: O6- Methylguanine-DNA Methyltransferase; VEGF: Vascular Endothelial Growth Factor; VEGFR: VEGF Receptor
Introduction
Glioblastoma Multiform (GBM) is one of the most aggressive malignancies of the central nervous system and it is considered among the deadliest human cancers [1]. The incidence rate of GBM is 3.1 per 100000 person-years and accounts for the 18.5% of all brain tumors, despite has increased slightly over the last decades, being more frequent among Caucasians [2,3]. The standard therapy for patients with GBM consists of surgery, fractioned radiotherapy with concomitant temozolamide, followed by adjuvant temozolamide. However, despite current available aggressive treatments, the prognosis for patients with GBM remains daunting, with a median survival time of 14.6 months and only 3.4% of patients remains alive after 5 years with treatment [4,5]. This low median survival of patients with GBM has been ascribed to de novo or acquired resistance to ionizing radiation [6]. Moreover, the high toxicity profile inherent to these therapies becomes poorly tolerated and often limits their minimum benefits [7]. Therefore, novel therapeutic paradigms are urgently required to overcome the inherent limitations of conventional treatments currently available for patients with GBM [8].
Most of high-grade gliomas overexpress the Epidermal Growth Factor Receptor (EGFR), which distinguish from the very low levels found in normal brain [9]. Mutation and gene amplification of the EGFR are associated with a more aggressive phenotype and a worse clinical outcome [10]. This has led to develop new molecular-targeted therapies based on the inhibition of this molecule. Among different strategies exploited to target the EGFR, monoclonal antibodies that bind directly extracellular epitopes of the receptor and smallmolecule tyrosine kinase inhibitors that inactivate the tyrosine kinase domain of the EGFR have undergone the more successful approaches so far in the clinic [11].
Nimotuzumab is a humanized monoclonal antibody against the EGFR that has shown promising results in clinical trials [12]. Nimotuzumab has undergone an extensive evaluation in GBM, showing efficacy as a single agent therapy or combined with ionizing radiation or chemotherapy [13-18]. The low toxicity profile of the antibody, together with its proven efficacy becomes nimotuzumab a promising therapeutic option for patients with GBM, especially in pediatric population [19]. Moreover, its ability to improve the efficacy of conventional cytotoxic therapies and the possibility to use used under long-term schemes without a dose-limiting toxicity highlights the need to elucidate the basic mechanisms by which this antibody acts [16,19].
Previous studies conducted by our group have corroborated the antitumor efficacy of nimotuzumab against U87MG xenografts either alone or in combination with ionizing radiation [20,21]. These studies have suggested that the antitumor activity of nimotuzumab in U87MG xenografts might be in part due to its ability to target the CD133+ subpopulation, in addition to effectively block the EGFR [20,21]. However, molecular determinants accounting for the ability of nimotuzumab to target the CD133+ cells remains unclear. In the current study we investigated whether U87MG cells with ability to form neurospheres express the CD133+ CSC molecular marker and whether CD133 is co-localized with EGFR protein as analyzed by immunofluorescence. Moreover, the growth-inhibitory effects of nimotuzumab in combination with ionizing radiation in U87MG cells were evaluated either in cell culture or growing as xenografts in NMRI nude mice.
Materials and Methods
Cells and tumor spheres culture
U87MG (HTB-14, ATCC) is a human GBM cell line. Cells were grown in a 1+1 mixture of Eagle’s MEM and Basal medium (Sigma) containing 2mM L-glutamine and 10% Fetal Bovine Serum (FBS) under a humidified atmosphere of 5%CO2 at 37°C. For obtaining tumor spheres, the cells were cultured as described previously [22] with modifications. The half of the medium was replaced with an equal volume of serum-free neural stem cell medium containing MEM, recombinant human epidermal growth factor (10 ng/ml; Sigma), and endothelial cell growth supplement factor (1μg/ml). This procedure was repeated every 24 h until several primary tumor spheres were visible under microscopy. At this point, all cell culture medium was discarded and maintained in serum-free neural stem cell medium. For obtaining secondary spheres, primary tumor spheres were mechanically disaggregated and cultured for additional 72 h in conditioned serum-free media MEM supplemented with EGF 10 ng/ mL and endothelial cell growth supplement factor 1 ug/mL.
Antibodies
The humanized anti-EGFR monoclonal antibody nimotuzumab was generated at the Center of Molecular Immunology [12]. Primary and secondary antibodies were purchased from commercial sources as listed: rat monoclonal anti CD31/PECAM-1 antibody (BD Pharmingen), horseradish peroxidase (HRP)-conjugated anti-rat IgG1 (Southern Biotech), biotin-conjugated mouse monoclonal antibody to CD133/1 (AC133) (Miltenyi Biotec), and HRPconjugated anti-mouse IgG (DakoCytomation).
Drug cytotoxicity assay
U87MG cells were seeded and plated onto 96-well plates at a density of 5x103 cells/well in 0.1 mL culture medium containing 10% FBS, and incubated at 37°C and 5% CO2. Twenty four hours later cells were exposed to different concentrations of nimotuzumab for the required time period. On days 0, 1, 2, and 3, culture medium was removed and 0.2 mL 3-(4,5)-dimethylthiahiazo(-z-yl)-3,5-diphenytetrazoliumromide (MTT) solution (0.5 mg/mL; Sigma) was added. The cells were incubated for 4h and the medium was replaced by 0.15 mL DMSO. The plates were agitated for 15 min and the optical density was measured at 550 nm in a photometer (Bio-TEK).
Immunofluoresence staining
For examination of neural stem cell markers, CD133 staining was detected with immunofluorescence. Immunofluorescence staining was performed following a protocol described [23] with modifications. The cells were deposed in slides, fixed in 4% paraformaldehyde, and frozen at -20°C until analysis. For immunostaining the cells were pretreated with normal goat serum for 30 minutes and then incubated with biotin-conjugated antibody against CD133/1 (1:10) for 1h, followed by incubation with streptavidine. For double labeling of CD133/EGFR, primary and secondary antibodies were used 1h for CD133 and overnight for EGFR. The cells were counterstained with DAPI to reveal the nuclei. The slides were examined and photographed with a laser confocal scanning microscope (Leica).
Immunohistochemistry
All the specimens were embedded in Tissue-Tek OCT (optimal cutting temperature) compound, shock frozen and stored in nitrogen until analysis. Immunostaining was performed on 4% paraformaldehyde-fixed, cryostat 5-μm tissue sections placed on glass slides. To detect microvessels, sections were stained with an antibody against CD31/PECAM-1 (1:100) as described previously [20]. Negative controls consisted of duplicate sections of the same specimens in which the primary antibody had been excluded and replaced with PBS or negative control immunoglobulin. Sections were visualized with 3,3’-diaminobenzidine as a chromogen and counterstained with Mayer’s hematoxylin. Representative tumor sections were identified on a light microscope (Zeiss, Axioskop 40) with an ocular magnification of X40 evaluating 4 to 5 tumors from each group.
Animal experiments
Female nude mice (8-10 weeks old, nu/nu) were obtained from Charles River. The mice were housed and maintained under aseptic conditions in facilities approved by the German Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the German Animal Protection Law, and their use was approved by the local responsible authorities. Animals met the requirements of the UKCCCR guidelines [24]. To produce xenografts, tumor cells were harvested from subconfluent cultures by treatment with 0.25% trypsin and 0.05% EDTA. Only single-cell suspensions with >90% viability were used for injections. Animals were inoculated with 107 U87MG tumor cells s.c. into left flank. Tumor volume were determined from direct measurement with calipers and calculated according to the formula: 0.5 x (large diameter) x (small diameter)2. Treatments were initiated three days after tumor cell injection. The treatment group consisted of eight mice, which received the anti-EGFR monoclonal antibody nimotuzumab concomitant with radiation. The antibody was administered intraperitoneal three times per week with 1 mg per mouse (50mg/ kg), during three weeks. For radiation, animals were exposed to a total dose of 3.0 Gy of total body radiation fractioned in 1.0 Gy weekly. The control group consisted of ten mice which received saline solution instead of the antibody. The antibody was administered 6 h before radiation therapy. All animals were sacrificed by day 36 when tumor weight from the control group exceeded the ten percent of total animal weight. Subcutaneous tumors were snapping frozen and stored for additional analyses.
Statistical Analysis
Statistical analysis was performed using the GraphPAD In-Stat software for Windows, version 4.0 (GraphPAD). Statistical testing was determined by Student´s t-test and P<0.05 was considered as significant.
Results
In vitro cytotoxicity of nimotuzumab in U87MG cells either alone or in combination with radiation
We first explored the cytotoxic activity of the anti-EGFR monoclonal antibody nimotuzumab alone or in combination with radiation on the growth of U87MG cell line. For that purposes U87MG cells were incubated with the antibody at different concentrations by 1, 2, or 3 days and its cytotoxic activity was assessed by MTT assay. Figure 1A shows the growth inhibition profiles upon antibody incubation at different time points. The maximal cytotoxic activity of nimotuzumab was reached at concentrations higher than 1 μg/mL, when cells were incubated with the antibody by 3 days. However, even under extreme conditions assessed (high antibody concentrations and large incubations) the cytotoxicity found in treated cells after antibody exposure was weak and non-significant compared to the control group.
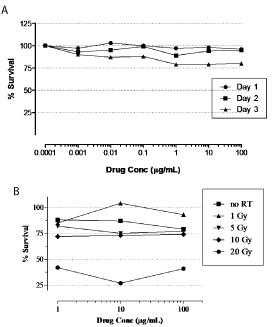
Figure 1: In vitro cytotoxicity of nimotuzumab alone or in combination
with radiation in U87MG tumor cells. In vitro cytotoxic activity of
nimotuzumab alone (A) or in combination with radiation (B) in U87MG tumor
cells was assessed by MTT assay. (A) U87MG tumor cells were seeded and
plated onto 96-well cell culture plates at a density of 5x103 cells/well and
left grown for 24 h. Then nimotuzumab was added to cell culture medium
at indicated concentrations and incubated for 1, 2, or 3 days. On day 4,
culture medium was removed and 0.2 ml 3-(4,5)-dimethylthiahiazol(-z-yl)-
3,5-diphenytetrazoliumromide (MTT) solution was added. The cells were
incubated for 4 h and the medium was replaced by 0.15 ml DMSO. The optical
density of the solution in the wells was measured at 550 nm in a photometer.
(B) For combination experiments, cells were irradiated with indicated doses
before seeded at Day 0.
Similar results were confirmed when the antibody was administered after the irradiation of the tumor cells (Figure1B). A significant cytotoxicity was only observed at the dose of 20-Gy of radiation, but it seems to be attributable to radiation-induced toxicity, according to the intrinsic radioresistance of U87MG cells previously reported [25]. These results suggest a weak in vitro cytotoxic activity of nimotuzumab alone or in combination with radiation in U87MG cells.
Nimotuzumab induces a significant tumor growth delay of U87MG xenografts injected subcutaneously in mice
The antitumor activity of nimotuzumab in combination with radiation therapy was further evaluated in U87MG xenografts in NMRI nude mice. U87MG cells were inoculated in the flanks of mice and animals were subsequently treated with nimotuzumab plus 1 Gy of radiation during 3 weeks. In contrast to in vitro results, the combination of nimotuzumab and ionizing radiation resulted in a substantial tumor growth delay and subsequent inhibition of the growth rate of U87MG xenografts compared to the control group (Figure 2). A maximal inhibitory effect on the growth of U87MG xenografts was found in mice treated with nimotuzumab plus radiation by day 30 after tumor inoculation compared to the control group. Tumor measurement at this time point revealed a half reduction in tumor size for those animals treated compared to the control group. This is a median relative tumor volume (set to 1 for the first measurement) for nimotuzumab plus radiation of 1.35 vs 2.85 for the control group. These results suggest that nimotuzumab had antitumor activity in combination with ionizing radiation in U87MG tumors in vivo under the dose and schedule proposed.
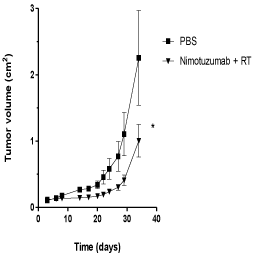
Figure 2: Antitumor activity of nimotuzumab and radiation in U87MG
cells subcutaneously xenografted into NMRI nude mice. Tumors were
formed by implanting 107 U87MG cells subcutaneously in nude mice (8 or
10 mice per group). Treatments were initiated 3 days after tumor inoculation
with nimotuzumab, 50 mg/kg intraperitoneally, three times per week by 3
weeks, and radiation (RT), 3Gy fractioned in 1Gy weekly. Mice in the control
group received PBS and served as a control. Tumor volume was determined
at the indicated time thereafter. Student´s t-test; symbols indicate statistical
differences.
Moreover, tumors formed in mice untreated exhibited hemorrhagic zones, with a widespread angiogenesis and a high density of microvessels, typically seen in human glioblastomas (Figure 3A, 3B, and 3E). In contrast, mice treated with nimotuzumab plus radiation showed tumors with a lower microvessel density and absence of hemorrhagic areas, suggesting an antiangiogenic activity for combination therapy (Figure 3C, 3D, and 3F). These preliminary findings were corroborated by CD31 immunostaining of tumor tissues from mice untreated (Figure 3G) or receiving the combination therapy (Figure 3H), confirming the antiangiogenic potential of nimotuzumab plus ionizing radiation in this tumor model.
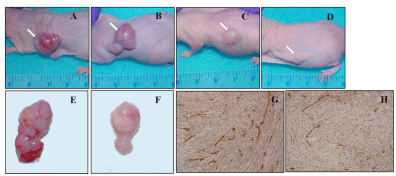
Figure 3: Subcutaneous tumorigenicity of U87MG tumors treated
with nimotuzumab and radiation. Tumors formed in untreated nude
mice by implanting with 107 U87MG cells showed glioblastoma multiformspecific
features (rapid growing tumors with highly vascularized areas
and surrounded by high density of microvessels) in the control group (A
and B), instead of nimotuzumab plus radiation treated animals (C and D).
Mice were sacrificed when tumor weight exceeded 10% of animal weight.
Representative tumor specimens from untreated (E) or nimotuzumab plus
radiation treatment (F) groups are shown. Untreated animals developed large
tumors with hemorrhagic masses whereas treated animals generated small
and poorly vascularized tumors. Immunohistochemical staining of frozen
sections of the tumors generated in the control group (E) and nimotuzumab
plus radiation group (F) are shown. Micro vessels were detected by staining
representative tumor sections with an antibody against CD31/PECAM-1
(brown). Subsequently, tumor blood vessels distribution was visualized under
light microscopy. White arrows indicate tumor tissues.
Generation of tumor sphere forming cells with selfrenewal capability in the U87MG cell line
Tumor sphere forming cells with tumorigenic properties have been described in GBM cell lines, being a primary source of radiation resistance [26]. We therefore investigated the capacity of U87MG cells to form non-adherent tumor spheres in fresh serum-free neural stem cell culture medium in presence of nimotuzumab. Based on the previous results obtained in the MTT assays, the maximal antibody concentration of 100 μg/ml of nimotuzumab was assessed in subsequent in vitro experiments. Many adherent non-sphere forming cells were observed in the bottom of the wells in culture plates seeded with 2x104 U87MG cells/well maintained in 10% FBS supplemented medium left untreated (Figure 4A) or treated with nimotuzumab (Figure 4B). In contrast, small visible spheres were generated after 7 days by progressively increasing serum-free neural stem cell medium either in medium alone (Figure 4C) or treated with nimotuzumab (Figure 4D). In 2 weeks, the diameter of these spheres progressively increased by 10- to 20-fold in both culture conditions (Figure 4E and 4F), suggesting that U87MG tumor sphere forming cells are also preferentially resistant to nimotuzumab in vitro.
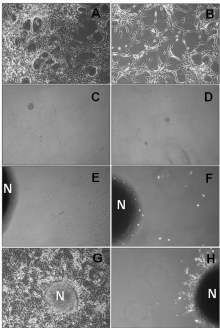
Figure 4: Generation of tumor sphere forming cells with self-renewal
capability in the U87MG cell line. U87MG cells were seeded on cell
culture plates and left grown in MEM supplemented with 10% FBS (A; B) or
serum-free neural stem cell medium containing MEM/F12, rhEGF, 20 ng/mL;
endothelial cell growth supplement factor 1 μg/mL (C - F) in the presence of
anti-EGFR monoclonal antibody nimotuzumab (100 μg/mL) (B; D; F) or left
untreated (A; C; E). Secondary neurospheres derived from single parental
cells from primary generated tumor spheres were recultured in medium
supplemented with 10% FBS by 72 hours in medium alone (G) or medium
containing nimotuzumab (100 μg/mL) (H). Representative microscope
images of culture plates are shown. N: neurospheres (20X).
Moreover, formed neurospheres were able to repopulate well in culture plates with adherent cells when culture medium was replaced by 10% FBS after 72 hours (Figure 4G). In contrast, only a few adherent non-sphere forming cells were visible to growth surrounding neurospheres when nimotuzumab was added to culture medium (Figure 4H).
CD133 and EGFR are co-localized in the membrane of tumor spheres from U87MG cells
CD133 is a cell surface marker for normal human neural stem cells widely accepted as a marker for brain CSC. We therefore compared CD133 expression in tumor sphere forming cells and adherent nonsphere forming cells in the U87MG cell line. As shown in Figure 5, CD133 staining was detected in tumor spheres elicited from U87MG cells (left panel), but not in adherent non-sphere forming cells (right panel). The EGFR cell membrane expression was previously corroborated in U87MG cells by FACS analysis (Supplementary Figure 1). Furthermore, formed neurospheres showed co-localized immunostained in the cell membranes for CD133 and EGFR proteins when cells were double stained (Figure 6), suggesting a potential molecular explanation by which nimotuzumab is able to target the CD133+ cells.
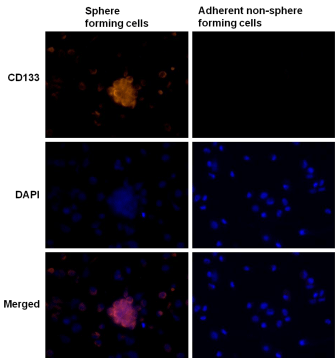
Figure 5: Expression of the neural stem cell marker CD133 in tumor
spheres forming cells. The expression of neural stem cell marker CD133
visualized under fluorescent microscopy evaluated in U87MG GBM spheres
arisen after culture in serum-free neural stem cell medium (left panel); or
adherent non-sphere forming cells growing in medium containing 10% FBS
(right panel). CD133 (red). DAPI (blue) reveals nuclei. (40X)
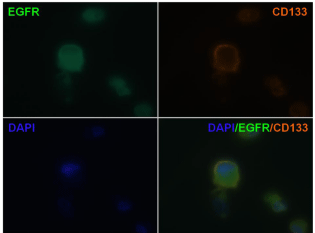
Figure 6: Co-localization of EGFR and neural stem cell marker CD133
on cells from secondary tumor spheres. The expression of EGFR and the
neural stem cell marker CD133 on cells from secondary tumor spheres was
visualized under fluorescent microscopy. EGFR (green); CD133 (red). DAPI
reveals nuclei (blue). (100X)
Discussion
GBM remains a deadly disease with a poor prognosis due to therapeutic resistance and frequent tumor recurrence after surgical removal. The disappointing results of standard therapy for GBM have led investigators to the search of new therapeutic approaches, in particular therapies based on well-defined molecular targets. One promising therapeutic target currently under investigation is the EGFR. Mutation and gene amplification of the EGFR have been associated with a more aggressive phenotype and a worse clinical outcome in recurrent GBM patients [10]. However, the clinical efficacy of EGFR antagonists in GBM is still limited, highlighting the need of a better understanding of the molecular mechanisms leading such antitumor efficacy.
We have previously demonstrated that nimotuzumab has a potent antitumor activity in combination with ionizing radiation in U87MG xenografts, acting as a radiosensitizing agent [20]. Moreover, nimotuzumab’s ability to target the CD133+ radioresistant population has been suggested to account, at least in part, for its antitumor activity in this tumor model. However, the molecular mechanisms by which the antibody is able to target the CD133+ cells are still unclear. By extending our previous findings, the current study proposes two hypotheses that could help to explain the ability of nimotuzumab to target the CD133+ cell population in U87MG tumor xenografts. One hypothesis relies on the ability of nimotuzumab to bind cells overexpressing surface EGFR, its natural target. Indeed, the current study demonstrated that CD133 molecule is co-localized with the EGFR protein on the cell surface of non-adherent spheres forming cells in the U87MG cell line, which is in line with a previous report indicating that CD133+ cells are contained in the EGFR+ cell population [27]. In such a case, the presence of the EGFR in the CD133+ subpopulation would be the enabling condition for nimotuzumab targeting, and the levels of expression of the molecule might dictate its sensibility to nimotuzumab regardless the expression of the CD133 molecule. Since several reports have linked the efficacy of nimotuzumab with the levels of expression of the EGFR in tumor tissues [28-30], further investigation will required to elucidate whether CD133+ subpopulation has a different level of expression of the molecule than CD133- cells, and therefore a differential threshold of sensibility to nimotuzumab, despite a preliminary report indicates no significant differences between the two populations [31]. Considering that previous studies have linked the resistance of brain tumor to therapies either with, EGFR overexpression or the presence of brain tumor stem cells, defined by the CD133+ population, it could be speculated that CD133+/EGFR+ cells may represent a particular resistant subpopulation, critical for developing a successful anticancer therapy.
In line with our results, other studies also reported EGFR coexpressed with the CD133 CSC marker in GBM cells [27,31-33]. Whereas Mazzoleni et al. classified different tumor-initiating populations of tumor cells in GBM based on the EGFR expression [27], another preliminary study on one GBM patient showed coexpression of CD133 and EGFR [33]. Moreover, previous findings from fresh brain tumor samples indicate that an effective blockade of the EGFR signaling reduces exogenous mitogen-independent GBM brain tumor stem cell proliferation, suggesting a key role for endogenous activation of EGFR and its signaling pathways [34]. However, in the previously mentioned study the abrogation of EGFR signaling did not eliminate exogenous EGF-independent GBM stem cell proliferation, suggesting that other mechanisms may also contribute to GBM cells survival and proliferation.
A second hypothesis associates the ability of nimotuzumab to target CD133+ cells with the sensibility of the CD133+ subpopulation to antiangiogenic therapies. Our results demonstrated that nimotuzumab had a significant antitumor efficacy against U87MG xenografts but a weak cytotoxic activity against U87MG cells in culture, even when combined with high doses of ionizing radiation. Despite the pertinence of in vivo operating mechanisms, such as antibody-induced complement activation, might help to explain differential efficacy in both scenarios, accumulated evidences suggest a key role for angiogenesis. Earlier studies in brain tumors have established a close and complex relationship between vessels and CD133+ niches, supporting the idea that brain tumor stem cells are located in perivascular niches with an intimate interplay between tumor stem cells and blood vessels [31,35,36]. Accordingly, the selective eradication of GBM CSC with antiangiogenic therapies by targeting the tumor microenvironment rather than CSC directly has been suggested [36,37]. In line with this hypothesis, antiangiogenic therapies may disrupt glioma CSC vascular niches leading a reduction or loss of brain stem cell characteristics, such as their increased capacity to repair DNA mismatch induced by cytotoxic drugs [35].
The molecular mechanism by which GBM stem cell-like cells promote angiogenesis has been linked to their capacity to secrete elevated levels of vascular endothelial growth factor (VEGF). A previous study suggested that CD133+ cells, but not CD133- cells, from human GBM patient specimens or human glioma xenografts were able to form highly angiogenic and hemorrhagic tumors when implanted either s.c. or intracranially in nude mice [35]. In such study VEGF levels in the CD133+ population were 10 – 20-fold than in CD133- cells modifying endothelial cell behavior. Moreover, the addition of bevacizumab, a neutralizing anti-VEGF antibody, suppressed the growth of xenografts derived from CD133+ cells, but showed a limited efficacy against xenografts derived from the CD133- population, suggesting the potential of VEGF neutralizing therapies to selectively eradicate CSC. In line with these findings, the antiangiogenic potential of nimotuzumab has been well demonstrated in different tumor models [20,21,38,39]. The mechanism by which nimotuzumab induces its antiangiogenic activity has precisely been linked to a potent downregulation of the VEGF secretion, which in turn may lead endothelial cell to apoptosis contributing to reduce neovascularity, as demonstrated in the squamous carcinoma cell line A431 [38].
Our in vitro experiments used specific culture conditions previously described to favor the presence of cells with the CSC phenotype in the U87MG cell line (e.g. CD133+ cells with enhanced capacity to form secondary neurospheres). We found that tumor cells in serum-free neural stem cell medium expressed the CD133 molecular marker, and when recultured in medium containing serum, tumor spheres became adherent and rapidly repopulated wells. Notwithstanding, our results must be interpreted with caution regarding the existence of a CSC population, since the confirmatory current standard for evaluating whether solid tumors contain CSC is a serial tumorigenic transplantation assay into immunodeficient animals [40].
Whether the capacity of specific agents to eradicate CD133+ cells may contribute significantly to their antitumor activity, despite the low frequency of this subpopulation within the tumor bulk, has been a hotspot of discussion over the last years. Controversy arises from the fact that the CD133+ subpopulation is very rare in human tumors and its frequency may fluctuate depending on culture conditions, which may help to explain disparate results between studies. In fact, the frequency of CD133+ cells in the U87MG cell line was reported lower than 0.2% in long-term cultures [41], whereas has found to decline with time in cell culture in human keratinocytes [42].
CD133+ CSC has been suggested the main source of resistance after chemo- and radiation therapy in GBM [26]. This has the premise to explain why such cytotoxic therapies can shrink a tumor, by eliminating non-CSC in the tumor bulk, but not eradicate CD133+ CSC, resulting in eventual tumor recurrence. As a consequence, improved rates of tumor responses or progression-free survival, but only negligible or no improvements in overall survival are frequently observed in treated patients. By contrast, it could be speculated that primarily eradication of CSC, which represent a minor faction of the overall tumor, by antiangiogenic therapies, might enhance the efficacy of cytotoxic therapies for prolonging overall survival in the absence of significant tumor shrinkages. Based upon such considerations, it might help to explain the clinical benefits of nimotuzumab in terms of response rate, control disease rate, and overall survival when the antibody has been administered to patients with GBM concomitant with chemo and/or radiation therapy in a number of clinical trials. One illustrative example is a randomized, phase III trial conducted in 149 newly diagnosed GBM patients that received nimotuzumab in addition or not to standard radiotherapy with temozolomide [18]. A stratified analysis within this study showed that patients with non-methylated MGMT (unresponsive to temozolomide) that received nimotuzumab had a median survival time of 19.6 months, as compared to 15.0 months for the same patients not receiving the antibody. Interestingly, CD133+ cells express higher levels of MGMT mRNA, compared to autologous CD133- cells, and such expression has been linked to the intrinsic GBM resistance to chemotherapeutic agents including temozolomide [43]. Moreover, these therapeutic outcomes highlight the importance of prospectively identifying nimotuzumab efficacy predictors in GBM, a hypothesis currently under evaluation in clinical trials.
In summary we demonstrated that nimotuzumab, a monoclonal antibody against the EGFR, inhibits the growth of U87MG xenografts when is administered in combination with ionizing radiation, despite its lack of cytotoxicity in vitro. This antitumor activity seems to be conditioned, at least in part, by a potent antiangiogenic response to the combinatorial therapy. The demonstrated presence of a CD133+/ EGFR+ subpopulation within the U87MG cell line with ability to growth in serum-free neural stem cell medium brings new insights about the molecular mechanisms by which nimotuzumab is able to target the CD133 radioresistant population, which may help to explain its radiosensitizing capacity against U87MG xenografts. Moreover, it illustrates well how a better comprehension of these mechanisms might impact in the design of clinical investigations to improve the therapeutic efficacy of the antibody in combination with standard cytotoxic therapies.
Acknowledgment
This work was partially supported by a grant from the UICC fellowship. Grant ICR/08/103 / 2008.We thank Monika Becker (Max Delbrück Center for Molecular Medicine) for technical assistance in animal experiments.
References
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114: 97-109.
- Dubrow R, Darefsky AS. Demographic variation in incidence of adult glioma by subtype, United States, 1992-2007. BMC Cancer. 2011; 11: 325.
- Fisher JL, Schwartzbaum JA, Wrensch M, Wiemels JL. Epidemiology of brain tumors. Neurol Clin. 2007; 25: 867-890, vii.
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008; 359: 492-507.
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10: 459-466.
- Feun LG, Savaraj N, Landy HJ. Drug resistance in brain tumors. J Neurooncol. 1994; 20: 165-176.
- Prados MD, Russo C. Chemotherapy of brain tumors. Semin Surg Oncol. 1998; 14: 88-95.
- Hauch H, Sajedi M, Wolff JE. Treatment arms summarizing analysis of 220 high-grade glioma studies. Anticancer Res. 2005; 25: 3585-3590.
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985; 313: 144-147.
- Lv S, Teugels E, Sadones J, De Brakeleer S, Duerinck J, Du Four S, et al. Correlation of EGFR, IDH1 and PTEN status with the outcome of patients with recurrent glioblastoma treated in a phase II clinical trial with the EGFRblocking monoclonal antibody cetuximab. Int J Oncol. 2012; 41: 1029-1035.
- Prados MD, Chang SM, Butowski N, DeBoer R, Parvataneni R, Carliner H, et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol. 2009; 27: 579-584.
- Mateo C, Moreno E, Amour K, Lombardero J, Harris W, Perez R. Humanization of a mouse monoclonal antibody that blocks the epidermal growth factor receptor: recovery of antagonistic activity. Immunotechnology. 1997; 3: 71-81.
- Cabanas R, Saurez G, Rios M, Alert J, Reyes A, Valdes J, et al. Treatment of children with high grade glioma with nimotuzumab: a 5-year institutional experience. MAbs. 2013; 5: 202-207.
- Solomn MT, Selva JC, Figueredo J, Vaquer J, Toledo C, Quintanal N, et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma in the treatment of high grade glioma patients. In. 2011.
- Bode U, Massimino M, Bach F, Zimmermann M, Khuhlaeva E, Westphal M, et al. Nimotuzumab treatment of malignant gliomas. Expert Opin Biol Ther. 2012; 12: 1649-1659.
- Bach F, Westphal M. Current status of a phase III trial of nimotuzumab (ti- EGF-R) in newly diagnosed glioblastoma. J Clin Oncol. 2011; 29.
- Fleischhack G, Siegler S, Warmuth-Metz M, Kortmann RD, Massimino M, Khuhlaeva E, et al. Final results of a phase III study consisting of nimotuzumab and concomitant standard radiotherapy for the treatment of newly diagnosed diffuse intrinsic pontine gliomas. Pediatr Blood Cancer. 2009; 755.
- Westphal M, Bach F. Final results of a randomized phase III trial of nimotuzumab for the treatment of newly diagnosed glioblastoma in addition to standard radiation and chemotherapy with temozolomide versus standard radiation and temoziolamide. In: ASCO Annual Meeting: J Clin Oncol. 2012; 30.
- Ramos TC, Figueredo J, Catala M, González S, Selva JC, Cruz TM, et al. Treatment of high-grade glioma patients with the humanized anti-epidermal growth factor receptor (EGFR) antibody h-R3: report from a phase I/II trial. Cancer Biol Ther. 2006; 5: 375-379.
- Diaz Miqueli A, Rolff J, Lemm M, Fichtner I, Perez R, Montero E. Radiosensitisation of U87MG brain tumours by anti-epidermal growth factor receptor monoclonal antibodies. Br J Cancer. 2009; 100: 950-958.
- Diaz A, Blanco R, Lemm M, Fichtner I, Leon K, Montero E. Preclinical efiicacy of nimotuzumab, an anti-EGFR monoclonal antibody as a single agent therapy in human GBM U87MG xenografts. J Cancer Ther. 2012; 3: 245-255.
- Yu SC, Ping YF, Yi L, Zhou ZH, Chen JH, Yao XH, et al. Isolation and characterization of cancer stem cells from a human glioblastoma cell line U87. Cancer Lett. 2008; 265: 124-134.
- Kabos P, Ehtesham M, Kabosova A, Black KL, Yu JS. Generation of neural progenitor cells from whole adult bone marrow. Exp Neurol. 2002; 178: 288- 293.
- Workman P, Twentyman P, Balkwill F, Blmain A, Chaplin D, Double J, et al. United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) Guidelines for the Welfare of Animals in Experimental Neoplasia (Second Edition). Br J Cancer. 1998; 77: 1-10.
- Li HF, Kim JS, Waldman T. Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiat Oncol. 2009; 4: 43.
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006; 444: 756-760.
- Mazzoleni S, Politi LS, Pala M, Cominelli M, Franzin A, Sergi Sergi L, et al. Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res. 2010; 70: 7500-7513.
- Ramakrishnan MS, Eswaraiah A, Crombet T, Piedra P, Saurez G, Iyer H, et al. Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. MAbs. 2009; 1: 41-48.
- Rojo F, Gracias E, Villena N, Cruz T, Corominas JM, Corradino I, et al. Pharmacodynamic trial of nimotuzumab in unresectable squamous cell carcinoma of the head and neck: a SENDO Foundation study. Clin Cancer Res. 2010; 16: 2474-2482.
- Garrido G, Tikhomirov IA, Rabasa A, Yang E, Gracia E, Iznaga N, et al. Bivalent binding by intermediate affinity of nimotuzumab: a contribution to explain antibody clinical profile. Cancer Biol Ther. 2011; 11: 373-382.
- Christensen K, Schrøder HD, Kristensen BW. CD133+ niches and single cells in glioblastoma have different phenotypes. J Neurooncol. 2011; 104: 129-143.
- Christensen K, Schrøder HD, Kristensen BW. CD133 identifies perivascular niches in grade II-IV astrocytomas. J Neurooncol. 2008; 90: 157-170.
- Schrot RJ, Ma JH, Greco CM, Arias AD, Angelastro JM. Organotypic distribution of stem cell markers in formalin-fixed brain harboring glioblastoma multiforme. J Neurooncol. 2007; 85: 149-157.
- Kelly JJ, Stechishin O, Chojnacki A, Lun X, Sun B, Senger DL, et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells. 2009; 27: 1722-1733.
- Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem celllike glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006; 66: 7843-7848.
- Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007; 11: 69-82.
- Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007; 67: 3560-3564.
- Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Bohlen P, Schlaeppi JM, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001; 61: 5090-5101.
- Crombet T, Osorio M, Cruz T, Roca C, del Castillo R, Mon R, et al. Use of the humanized anti-epidermal growth factor receptor monoclonal antibody h-R3 in combination with radiotherapy in the treatment of locally advanced head and neck cancer patients. J Clin Oncol. 2004; 22: 1646-1654.
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006; 66: 9339-9344.
- Wu A, Oh S, Wiesner SM, Ericson K, Chen L, Hall WA, et al. Persistence of CD133+ cells in human and mouse glioma cell lines: detailed characterization of GL261 glioma cells with cancer stem cell-like properties. Stem Cells Dev. 2008; 17: 173-184.
- Perrella G, Brusini P, Spelat R, Hossain P, Hopkinson A, Dua HS. Expression of haematopoietic stem cell markers, CD133 and CD34 on human corneal keratocytes. Br J Ophthalmol. 2007; 91: 94-99.
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006; 5: 67.