
Research Article
J Stem Cell Res Transplant. 2014;1(2): 1009.
Trichostatin A Induces Both Cell Division Arrest and Neural-linage Differentiation of the Mesenchymal Stem Cells Possibly by Triggering Wnt/β-Catenin Signaling
Bei-Yu Chen1, Meng-Meng Liang2†, Mo-Han Dong3†, Jie-Qiong Zhang3, Jing-Jie Wang4, Mo Li1, Xi Wang2, Zhuo-Jing Luo1* and Liang-Wei Chen2*
1Department of Orthopedics, Xijing Hospital, The Fourth Military Medical University, Xi’an 710032, China
2Institute of Neurosciences, The Fourth Military Medical University, Xi’an 710032, China
3Department of Military & Prevention Medicine, Fourth Military Medical University, Xi’an 710032, China
4Department of Gastroenterology, Tangdou Hospital, The Fourth Military Medical University, Xi’an 710038, China
†These authors have equal contribution to this work
*Corresponding author: Liang-Wei Chen, PhD, Institute of Neurosciences, The Fourth Military Medical University, Xi’an, 710032, P.R. China
*Corresponding author: Z.J. LUO, Department of Orthopedics, Xijing Hospital, The Fourth Military Medical University, China
Received: August 02, 2014; Accepted: September 27, 2014; Published: September 29, 2014
Abstract
Introduction: The bone marrow-derived mesenchymal stem cells (BM-MSC), with active proliferation and pluripotent differentiation abilities, present an ideal source of cell transplant therapy. It remains obscure, but, how to promote their neural-linage differentiation efficiently and controlling moderate proliferation for clinical application in neurological diseases.
Methods: By in vitro culture of rat BM-MSC, regulation of a histone deacetylase inhibitor trichostatin A (TSA) in both cell proliferation and neural-linage cell commitment, and possible involvement of Wnt/β-catenin signaling pathway in TSA-inducing biological effect were examined in this study.
Results: By cell count and flowcytometry, TSA exhibited cell division or cycle arrest of BM-MSC at doses above 10ng/ml by increased percentage of G1/S phase and apoptotic cells. Immunocytochemistry and immunoblot revealed promotion effect of TSA on neuronal cell differentiation with appearance of neuron-like ones and increased nestin and Tuj-1 expression in BM-MSC treated with TSA at dose of 100 and 500ng/ml. Enhanced glial cell differentiation was also observed by upregulation of NG2, glial fibrillary acidic protein and CNPase expression in BM-MSC with TSA treatment dose-dependently. Furthermore, significant activation of Wnt/β-catenin signaling was detected in TSA-treated BM-MSC, while Wnt signaling inhibitor IWR1 impeded above TSA-induced effects on BM-MSC.
Conclusion: This study has provided new evidence that TSA could induce neural-linage differentiation and cell division arrest of BM-MSC dose-dependently by triggering Wnt/β-catenin signaling activation, suggesting that TSA may be applied as a candidate drug in manipulation of MSC therapy for the treatment of various neurological disorders.
Keywords: Bone marrow-derived mesenchymal stem cells; Trichostatin A; Histone deacetylase; Wnt/β-catenin signaling; Cell transplant therapy
Abbreviations
BM-MSC: Bone Marrow-derived Mesenchymal Stem Cells; GFAP: Glial Fibrillary Acidic Protein; TSA: Trichostatin A
Introduction
Growing studies have shown that the bone marrow-derived mesenchymal stem cells or mesenchymal stromal cells (BM-MSC) may present an ideal source for tissue engineering or cell transplant therapies in human beings [1,2]. While wide heterogeneity was recognized in BM-MSC depending on their origins, the International Society for Cellular Therapy in 2006 proposed three minimal criteria for cultured human MSC definition [3]: i) when maintained in standard culture conditions, MSC must be plastic-adherent. ii) MSC must express CD73, CD90 and CD105, and lack expression of CD34, CD45, CD14 or CD11b, CD79 or CD19 and HLA-DR surface molecules, and iii) MSC must in vitro differentiate to osteoblasts, adipocytes and chondroblasts. Besides, BM-MSC can also be committed into cell lineages such as neural precursors, cardiomyocytes, liver cells, and possible other cell types. In fact, fast increasing of MSC knowledge and techniques has already led to some clinical trials for therapeutic manipulation of bone and cartilage diseases [4]. However, neural cell fate commitment or induced-differentiation mechanism of BM-MSC still remains a big clinical obstacle regarding their translational purpose for neurological disorders. Inspiringly, it is expected that effective production of MSC-derived functional neural-linage cells shall further extend their therapeutic application in various neurodegenerative diseases and nerve injury events [4].
Obviously, it is critical that efficiency of neural cell inducement and moderate control of cell proliferation are secured for establishment of a practical and safe MSC therapy [2,4]. Though a number of substances, neurotrophic or growth factors were found to exhibit certain promoting effect on neural cell differentiation, tumorigenicity of pluripotent stem cells still remains one most concern issue in cell transplantation [4]. Indeed, currently, there are still lacked of well-defined factors, drugs or chemicals with such both natural properties, i.e. neural cell fate commitment-promoting benefit and tumorigenicity-blocking effects in BM-MSC [4]. Recently, evidences indicated that certain epigenetic factors were actively involved in regulating differentiation of embryonic or adult stem cells [5-8]. For instance, Trichostatin A (TSA), a histone deacetylation inhibitor previously used for an anti-cancer agent, could sufficiently induce neuronal cell fate from the embryonic neural stem cells [9]. Moreover, only these embryonic neural stem cells with TSA treatment could definitely differentiate into functional neurons that were characterized with active potential and induced spikes, and underling mystery or inducing mechanism attracts a great attention [9]. In this study, interestingly, we demonstrated that TSA could effectively induce both neural-linage cell commitment and cell division or cycle arrest of rat BM-MSC, possibly through triggering Wnt/β-catenin signaling activation by using cell culture, flowcytometry, immunocytochemistry and western blot methods.
Materials and Methods
A total of twenty adult female Sprague-Dawley (SD) rats were used in the present study and supplied from the Animal Center of the Fourth Military Medical University of China. All animal experiments followed were carried out in according with the National Institute of Health guide for the care and use of Laboratory animals (NIH Publications No. 80-23) revised 1996, approved by the Committee of Animal Use for Research and Education of the Fourth Military Medical University, and all efforts were made to minimize animal suffering and reduce the number of animals used.
For cell culture preparation of BM-MSC, bone marrow mesenchymal cells were isolated from the femur bone of rats, which were sacrificed by animal decapitation. The bone marrow were washed out in PBS at room temperature, collected in glass tube and allowed to precipitation for a while. The cell suspension was seeded in dish in 10ml of MEM-α medium (Invitrogen) supplemented with 15% fetal bovine serum (Biosera, UK) and allowed to grow for 4-6 days until cell clones appeared. Cell pellets were collected, re-suspended and seeded in density 0.5-1×106 in T25 flasks (Corning) in MEM growth medium. After culture in a humidified 5% CO2/95% air incubator at 37°C about 5-7 days and cells grew fast in attached growth mode. They were dissociated by using accutase digestion and subjected to 5-6 passages each in about 3-5 days. After immunostaining confirmation of specific biomarkers (CD90+/CD45¯), BM-MSC cultured in 5-6 passages were used for following TSA experiments.
TSA treatment was performed to detect its influence on cell proliferation and cell differentiation of BM-MSC in vitro. Cell cultures of MSC were prepared in these groups, i.e. pre-treat, control, TSA-1ng/ml, 10ng/ml, 50ng/ml, 100ng/ml, 500ng/ml, 1000ng/ml and 2000ng/ml working dilutions. TSA (Sigma-aldrich, St Louis, MO, 78552), which was dissolved in DMSO and diluted with PBS, was added in cell culture medium, incubated with DMEM/F12 (Gibco) medium supplemented with 2% B27 (Invitrogen) without bovine serum and continued for 3 days and 7 days. Cell survival and cell growth ware observed firstly under phase-contrast microscopy. MSC cell samples of control, TSA-10ng/ml, 100ng/ml and 500ng/ml group of 3d or 7d time-points were freshly collected for flowcytometry and western blot, or the cultured cells were terminated by fixation with 4% paraformaldehyde for 10 min for immunocytochemical detection of morphology and growth state of differentiated MSC.
Flowcytometry
Flowcytometry was performed to detect cell cycle, cell viability and apoptosis of cultured BM-MSC in a standard protocol. Fresh cell samples were collected and centrifuged for 5min and supernatants were discarded. Cell pellets were suspended and incubated with 1-2ml accutase for 10 min at 37°C. After gentle repeated beating, cell suspension was re-centrifugated for 5min at 1000r, and supernatant was abandoned. The cells were re-suspended in 1ml PBS supplemented with 2ml dehydrated alcohol, shaken for a moment, sealed and kept at 4°C overnight. The prepared cell samples of BM-MSC were measured under a flowcytometer for cell cycle, cell survival, necrosis and apoptosis. For data presentation, percentages of defined cells like G1, S, and G2 phase, survival, necrosis, early apoptosis or late apoptosis were quantified among control, TSA-10ng/ml, 100ng/ml and 500ng/ ml groups.
Immunocytochemistry
Immunofluorescent staining was performed to demonstrate neural cell markers and Wnt/β-catenin signaling molecules in cultured BM-MSC. After culture for 3d and 7d in 24-well plates, cells were processed for immunocytochemical staining protocol. Briefly, cells were incubated with 10% donkey serum-containing blocking solution for 30min at room temperature, and followed by incubation of primary antibody solution containing 10% donkey serum, 0.3% triton X-100 in PBS at 4°C for 24h, i.e. rabbit anti-nestin (Sigma-aldrich, 1:1000), rabbit anti-β-tubulin III (Tuj-1, Sigma-aldrich, 1:500), rabbit anti-NG2 (Sigma-aldrich, 1:800), mouse anti-glial fibrillary acidic protein (GFAP, Dark, 1:4000) and rabbit anti-CNPase (Sigma-aldrich, 1:500), respectively. After three washes with PBS, cells were followed with incubation of Alexa Fluor-488, or Alexa Fluor-594 conjugated donkey anti-mouse or rabbit IgG (1:500, Molecular Probes) for 4h at room temperature. Finally, nuclear counterstaining with DAPI (Sigma-aldrich, D9564) for 10min was performed to visualize total cultured MSC. After PBS wash, cell samples were mounted with Fluorescence-preserving VECTASHIELD Mounting medium (Vector, H-1000) and examined under epifluorescent microscope and laser scanning confocal microscope (LSCM, Olympus, FV-1000). For control staining experiment, the primary antibody was substituted with normal mouse or rabbit serum. Immunoreactive cells were not detected in controls (data not shown), and specificity of antibodies used was also provided by manufacture’ data sheet.
Western blot
Western blot was performed to quantify expression of several neural cell markers and Wnt/β-catenin signaling molecules in cell culture in a standard protocol. Briefly, protein extracts were prepared from cultured MSC. Fresh cell samples were collected and homogenized at 4°C in 5 volumes of ice-cold lysis buffer [50mM Tris (pH 7.4), 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail (Complete, Roche Diagnostics)], and centrifugation of homogenates for 10 min (12000g) was then performed. After protein concentration was determined by the BCA assay (Beyotime, China), supernatant was mixed with four volumes of protein loading buffer, boiled for 5min at 99°C and stored at 4°C. Total protein (20-30 μg per loading passage) was loaded using loading buffer commercial for electrophoresis on 10-12% SDS-PAGE gels and transferred to the nitrocellulose membranes (Bio-Rad). After these membranes were blocked with 5% skimmed milk (Wandashan, China) in Tris-buffered saline containing 0.05% Tween 20, immunoblotting was performed with primary antibody to nestin, Tuj-1, NG2, CNPase, GFAP, Wnt1, Wnt3a, Wnt5a, β-catenin or phosphor-β-catenin respectively, and followed by secondary antibody incubation. Mouse anti-Wnt1 (Sigma-aldrich, 1:1000), rabbit anti- Wnt3a (abcam, 1:500), mouse anti-Wnt5a (Sigma-aldrich,1:1000), rabbit anti-β-catenin (Sigma-aldrich, 1:1000), rabbit anti-phosphor- β-catenin (Sigma-aldrich, 1:1000), and rabbit anti-β-actin (Sigma-aldrich, 1:800) were used. Visualization and detection of immunoblot bands was performed by an enhanced chemiluminescence (ECL) detection system (CWBio, China), and images were digitally acquired using ChemiScope System (Clinx, China). By using β-actin as internal control, quantitative analysis was carried out and data were presented in ratios.
Wnt signaling inhibition experiment
To confirm role of Wnt/β-catenin signaling pathway in TSA-induced cell differentiation of MSC, a blocking experiment was carried out in cell culture by administration of a specific inhibitor IWR-1, which can induce stabilization of APC/Axin2/GSK3β complex and decrease of cytoplamic free β-catenin level through a direct interaction and works as Wnt signaling inhibitor [10,11]. In TSA+IWR1 group, IWR-1 (Sigma-aldrich, I0161) was dissolved in saline and added in culture medium in 10 μM at 10min before TSA treatment (TSA-100ng/ml plus IWR1), and saline group was used as controls. After maintaining culture for 3d and 7d, differentiation cell markers and changes were examined in BM-MSC of control, TSA-10, 100, 500ng/ml, and TSA-100ng/ml+IWR1-10 μM group by western blot and immunocytochemistry.
Statistic analysis
For data analysis of flowcytometry, immunocytochemistry and western blot, total MSC cells, immunopositive cells and immunoblot density of nestin, Tuj-1, NG2, GFAP, CNPase, Wnt1, Wnt3a, Wnt5a, phosphor-β-catenin and β-catenin were counted or measured and given as mean ± S.E.M. (n=3-5, independent experiments). Differences between means were analyzed by one-way ANOVA (SPSS 18.0) with different markers or signaling molecules as independent factors. When ANOVA showed significant difference among means, the pair-wise comparisons between means were also performed by post hoc testing, and the significance level was set at a P value of 0.05 for all analyses.
Results
Cell division arrest and cell apoptosis of BM-MSC induced by TSA treatment
Influence of TSA treatment on cell survival, apoptosis and cell cycle were first observed in cell culture after BM-MSC in high-purity and uniform were established through 5-6 passages and characterized with specific BM-MSC biomarkers as reported previously [3,4]. Cell morphology and cell density of BM-MSC were observed and compared among these groups, i.e. pre-treat, control, TSA-1ng/ml, 10ng/ml, 50ng/ml, 100ng/ml, 500ng/ml, 1000ng/ml and 2000ng/ml (Figure 1A). Obvious changes in morphology and cell numbers of BM-MSC dose-dependently occurred at doses of above TSA-50ng/ ml working dilution. Cell count data revealed that BM-MSC in single unit field decreased after treatment of TSA dose-dependently and significantly in comparison with that of control (Figure 1B).
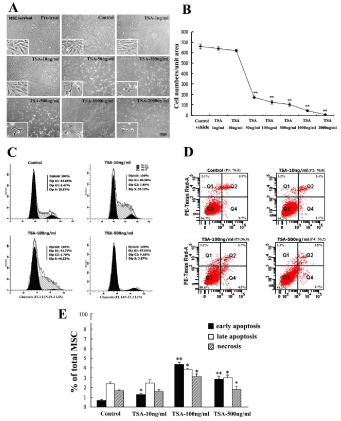
Figure 1: Influence of Trichostatin A (TSA) on cell survival, cell cycle and cell apoptosis of rat BM-MSC. A, Morphology and density of cultured BMMSC, i.e. groups of pretreat, control, TSA-1ng/ml, 10ng/ml, 50ng/ml, 100ng/ ml, 500ng/ml, 1000ng/ml and 2000ng/ml working dilutions; B, Quantitative comparison of BM-MSC survival cells among above groups; C, Cell cycle of BM-MSC in control, TSA-10ng/ml, 100ng/ml and 500ng/ml groups; D, Cell survival, necrosis and apoptosis pools of BM-MSC in control, TSA-10ng/ ml, 100ng/ml and 500ng/ml groups; E, Comparison of necrosis, early and late apoptosis of BM-MSC in percentages among above groups. ANOVA: *P<0.05, **P<0.01 vs control (Mean±SEM, n=3-5).
Flowcytometry analysis was performed to quantify cell survival, cell division and cell apoptosis of BM-MSC in control, TSA-10ng/ ml, 100ng/ml and 500ng/ml group. Cell cycle analysis indicated cell division or cycle arrest occurred after treatment of TSA, and increased percentages of cells at G1 and S phase were detected in BM-MSC treated with TSA-100ng/ml and 500ng/ml (Figure 1C). The percentage of apoptotic cells also increased in BM-MSC with treatment of TSA- 100ng/ml and 500ng/ml (Figure 1D). The percentages of necrosis, early and later apoptotic cells were quantified among control and TSA groups (Figure 1E). Quantitative data analysis indicated that TSA-induced decreasing of BM-MSC might be resulted from both cell cycle arrest and cell apoptosis.
Increased neuronal-like cell differentiation of BM-MSC induced by TSA treatment
Interestingly, promoting effects of TSA treatment on neuronal cell differentiation were observed and indicated by enhancing expression levels of neuronal cell markers and appearance of neuron-like cells in the cultured BM-MSC. Percentages of nest in (neural stem cell marker) and Tuj-1 (neuronal cell marker)-immunopositive ones of total MSC cells increased at dose of TSA-100ng/ml and 500ng/ml groups. Besides, a number of neuron-like cells appeared and grew thin processes in TSA-100ng/ml and 500ng/ml groups (Figure 2A). Quantitative data analysis showed increased percentages of nestin and Tuj-1-immunopositive ones constituting total MSC significantly at TSA-100ng/ml and 500ng/ml in comparison with that of control (Figure 2B).
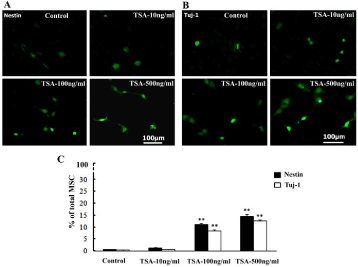
Figure 2: Increasing neuronal cell differentiation of cultured BM-MSC induced by TSA treatment. A-B, Nestin and Tuj-1-immunopositive cells in BM-MSC of control, TSA-10ng/ml, 100ng/ml and 500ng/ml groups at 3d culture; C, Comparison of nestin and Tuj-1-immunopositve cells among above groups in the percentage (%) of total DAPI-counterstained (not shown) BM-MSC. ANOVA: **P<0.01 vs control (Mean±SEM, n=5).
Enhanced glial cell differentiation of BM-MSC induced by TSA treatment
The stimulating effect of TSA treatment on glial cell differentiation were also observed in cell culture of BM-MSC, which was indicated by increasing expression of glial cell marker NG2, GFAP and CNPase. The NG2, GFAP and CNPase-immunopositive cells were observed in all groups, i.e. control, TSA-10ng/ml, 100ng/ml and 500ng/ml. However, stronger immunoreactivity to NG2, GFAP or CNPase was detected after TSA treatment and obviously at TSA-100ng/ml and 500ng/ml groups (Figure 3A). Cell count showed that increased percentages of NG2, GFAP and CNPase-immunpositive ones constituting total MSC occurred at TSA-100ng/ml and 500ng/ml group in comparison with that of control (Figure 3B).
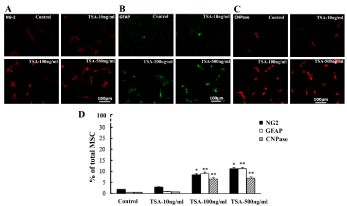
Figure 3: Enhancing glial cell differentiation of cultured BM-MSC induced by TSA treatment. A-C, NG2, GFAP and CNPase-immunopositive cells in BMMSC of control, TSA-10ng/ml, 100ng/ml and 500ng/ml groups at 3d culture; D, Comparison of NG2, GFAP and CNPase-immunopositive cells among above groups in the percentages (%) of total DAPI-counterstained (not shown) BM-MSC. ANOVA: * P<0.05, **P<0.01 vs control (Mean±SEM, n=5).
Activation of Wnt/beta-catenin signaling pathway in BMMSC with TSA treatment
Western blot was applied to examine possible role of Wnt/β- catenin signaling pathway in TSA-induced differentiation of BM-MSC at 3d and 7d culture. It revealed increase of total β-catenin and decrease of phosphor-β-catenin, the key molecules of Wnt/β-catenin signaling pathway (Figure 4A and 4E). Western blot also showed that expression level of total β-catenin molecule at 3d culture was more obvious in comparison with that of β-catenin at 7d culture. Quantification showed up-regulation of β-catenin proteins and down-regulation of phosphor-β-catenin significantly at dose of TSA- 100ng/ml and 500ng/ml in comparison with control, indicating that TSA could induce upregulation of active form of β-catenin molecule (Figure 4B and 4F). Besides, expression of Wnt1, Wnt3a and Wnt5a was also observed in cultured BM-MSC with TSA treatment, and data indicated increasing levels of Wnt1 and Wnt5a but not Wnt3a in cultured MSC after TSA treatment (Figure 4C and 4D).
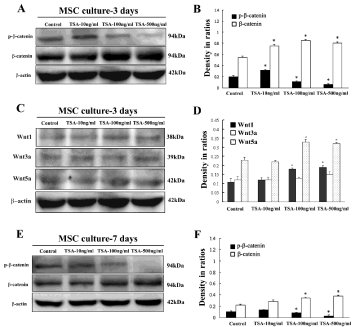
Figure 4: Expression levels of Wnt/β-catenin signaling molecules in cultured BM-MSC with or without TSA treatment. A, Expression of phosphor-β-catenin and total β-catenin of MSC of control, TSA-10 ng/ml, 100ng/ml and 500ng/ ml groups at 3d culture; B, Comparison of phosphor-β-catenin and total β-catenin among above groups in ratios to β-actin; C, Expression of Wnt1, 3a and 5a of MSC of control, TSA-10ng/ml, 100ng/ml and 500ng/ml groups; D, Comparison of Wnt1, 3a and 5a among above groups; E, Expression of phosphor-β-catenin and total β-catenin of MSC of control, TSA-10ng/ml, 100ng/ml and 500ng/ml groups at 7d culture; F, Comparison of phosphor- β-catenin and β-catenin among above groups in ratios to β-actin. ANOVA: * P<0.05, **P<0.01 vs control (Mean±SEM, n=3).
Wnt signaling inhibition experiment was performed to confirm involvement of Wnt/β-catenin signaling activation in TSA-inducing MSC differentiation by administration of a specific Wnt signaling blocker IWR1 [10, 11]. Western blot quantified relative levels of nestin, Tuj-1, NG2, GFAP and CNPase of cultured MSC among distinct groups of control, TSA-10ng/ml, 100ng/ml, 500ng/ml, TSA- 100ng/ml+IWR1-10μM. Increasing of nestin, Tuj-1, NG2, GFAP and CNPase-immunoblot bands were observed in TSA-treated MSC groups (Figure 5A and 5C). Quantitative analysis confirmed that significant upregulation of nestin, Tuj-1, NG2, GFAP and CNPase expression occurred in cultured MSC in TSA groups in comparison with that of control in ratio to internal control β-actin (Figure 5B and 5D). On the other hand, expression of nestin, Tuj-1, NG2, GFAP and CNPase decreased significantly in TSA+IWR1 group, and these neuron-like or glia-like cells also went down obviously or almost disappeared in this TSA+IWR1 group (data not shown).
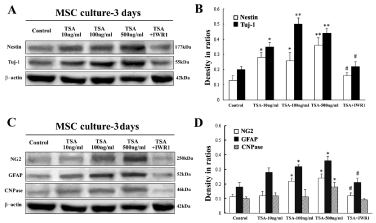
Figure 5: Expression of neural-linage cell markers, i.e. nestin, Tuj-1, NG2, GFAP and CNPase in cultured BM-MSC with or without TSA treatment. A, Expression of nestin and Tuj-1 in BM-MSC of control, TSA-10ng/ml, 100ng/ ml, 500ng/ml, and TSA-100ng/ml+IWR1 group at 3d culture; B, Comparison of nestin and Tuj-1 expression levels among above groups in density ratios to β-actin; C, Expression of NG2, GFAP and CNPase in BM-MSC of above groups; D, Comparison of NG2, GFAP and CNPase expression levels among above groups in ratios to β-actin. ANOVA: * P<0.05, **P<0.01 vs control; # P<0.05 vs TSA-100ng/ml group (Mean±SEM, n=3).
Appearance of TSA-induced differentiated cells in prolonged BM-MSC culture
Finally, TSA-induced differentiated cells were observed at 3d and 7d MSC culture. Although Tuj-1, GFAP, and CNPase-positive cells seemed similar appearance at 3d culture, these differentiated MSC ones showed much different morphology at prolonged 7d culture. These cells with immunoreactivity to Tuj-1 (neuronal cell marker), GFAP (astrocytic cell marker), and CNPase (oligodendrocytic cell marker) are shown (Figure 6A). Besides, percentages of Tuj-1, GFAP, or CNPase-immunopositive ones constituting total MSC cells (DAPI counterstained) were counted and shown (Figure 6B). Data indicated that a small population of MSC was induced into Tuj-1, GFAP, or CNPase-immunopositive cell commitment in both 3d and 7d culture of MSC of TSA-500ng/ml groups.
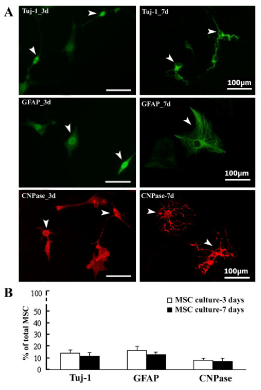
Figure 6: The prolonged neural-linage cell differentiation and maturation of cultured BM-MSC induced by TSA treatment. A, Morphological appearance of Tuj-1, GFAP, CNPase-immunopositive differentiated cells in BM-MSC with treatment of TSA-500ng/ml at both 3d and 7d culture; B, Comparison of Tuj- 1, GFAP, CNPase-immunopositive cells in percentages (%) of total BM-MSC between 3d and 7d.
Discussion
This study revealed that TSA promoted neural-linage cell differentiation of rat BM-MSC and property of differentiated cells was indicated by morphological evaluation and differentiation marker analysis. On the other hand, TSA also induced cell division or cell cycle arrest of BM-MSC dose-dependently. More interestingly, this study demonstrated potential involvement of Wnt/β-catenin signaling pathway in TSA-induced neural cell differentiation of BM-MSC. Further extensive studies should be needed to show functional property or maturation of the MSC-derived neuronal and glial cells. Nevertheless, this study has provided new evidence that TSA could effectively induce neural-linage commitment of BM-MSC possibly by triggering Wnt/β-catenin signaling pathway, suggesting that TSA may be applied as one candidate for MSC transplant therapy for various neurological diseases and nerve injury [2,4].
Neural cell fate decision of BM-MSC might be induced by TSA, a histone deacetylation inhibitor Growing studies have focused on epigenetic modulation of stem cell proliferation and differentiation, in which DNA histone acetylation, demethylation, microRNA regulation modes might be actively involved the forebrain neurogenesis [12], hippocampal neurogenesis [13-16], and cell differentiation of oligodendrocyte progenitors [17-20]. Potential interaction of epigenetic factors with growth factor or retinoic acid might occur in the neuronal differentiation [21,22]. Consistently, Balasubramaniyan et al reported interesting result of TSA-inducing neuronal differentiation from neural precursor cells, and only these progenitors with pretreatment of TSA could differentiate into neuronal cells [9]. It showed that TSA not only induced neuronal cell differentiation, but also fostered neuronal maturation, dendrite growth and spikes of new-born or differentiated neurons [9]. Although it still remains unclear how TSA could improve differentiation or trans-differentiation of MSC, previous studies identified that several types of agents could increase efficiency of cell reprogramming induced by defined factors and nuclear transfer. These agents include histone deacetylase inhibitor TSA and valporic acid, and other modulators of cell specific signaling, which might confer enhancing expression of several genes, cell plasticity and pluripotency [23,24].
On other hand, it was noticed that chemically-induced MSC cells might adopt some molecular properties of neuronal-like cells but did not have basic neuronal functional activities [25]. While these MSC differentiated using chemical inducers such as β-mercaptoethanol showed increases in expression of some neuronal markers, they did not show electrophysiological properties of neurons [25]. Liu et al also reported that morphological changes and increases in immunolabelling for certain cellular markers might be likely a result of cytotoxicity, cell shrinkage and changes in cytoskeleton [26]. Other authors suspected that neural trans-differentiation of bone marrow stromal cells induced by chemical agents might also be a short-time reversible phenomenon [27]. In our study, DMSO was also used and remained same low concentrations in both control and all TSA groups, and data indicated that increase of differentiated MSC might be a result from specific TSA intervention effect instead of DMSO toxicity. However, we can still not completely exclude the possibility that TSA-induced MSC differentiation might result in abnormal epigenetic changes as compared to normal phenotype.
Low tumorigenicity of BM-MSC treated with TSA may be beneficial for clinical therapy In the cell therapy for clinical application, tumorigenicity of transplanted MSC or pluripotent stem cells raises great concern and present one big hurdle in safety [28]. Presently, no a practical method can completely eliminate tumorigenicity of stem cells including BM-MSC, and an alternative may be to use differentiated neurons [29,30]. It is well known that tumorigenicity of transplanted stem cells was greatly influenced by recipients’ immune condition or response [31]. Studies have also shown that MSC exhibit immunosuppressive capacity and modulate immune function of cell populations including antigen presenting cells, T cells, B cells and natural killer cells [32,33]. Although underlying mechanisms of immunosuppressive effects of MSC or how MSC inhibit the proliferation of a variety of immune cells are still obscure, recent more evidences have suggested that MSC might down-regulate T and B lymphocytes, natural killer cells and antigen presenting cells through cell-cell interactions and soluble factor production [33-36].
A sharp decrease of MSC numbers was observed between TSA- 10ng/ml and 50ng/ml treatments in this study, suggesting that effective inhibition of TSA on MSC survival or growth occurred at dose above 10ng/ml. Flowcytometry analysis showed obvious cell division or cycle arrest of BM-MSC after treatment of TSA at 100ng/ml and 500ng/ml working dilution. Besides, apparent cytotoxicity of TSA was observed at doses of 1000ng/ml and 2000ng/ml. Data of this study suggested that doses of 50-500ng/ml might be suitable for TSA intervention for purpose of promoting neural-linage differentiation and controlling moderate proliferation of BM-MSC. While the immunosuppressive effect of MSC may foster tumorigenicity of transplanted stem cells, BM-MSC pretreated with TSA and characterized with controlling cell proliferation and promoting cell differentiation may be alternative choice in practical transplantation therapy [32].
Wnt/β-catenin signaling might be involved in regulating both neural-linage cell commitment and cell proliferation of BM-MSC Although TSA is clearly identified as a histone deacetylase inhibitor functionally, detailed down-stream signaling underling MSC differentiation remains obscure [4]. The Wnt signaling functions as a key regulating signal for stem cell proliferation and cell differentiation [37,38]. In this study, western blot analysis indicated treatment of TSA at 100ng/ml and 500ng/ml decreased p-β-catenin expression levels of MSC. By comparison with total β-catenin level, it indicated activation of wen/β-catenin signaling in MSC after treatment of TSA above dose of 100ng/ml. Several studies showed that Wnt signaling influenced proliferation and differentiation of human MSC and osteogenic process [39-42]. Regard et al reported that Wnt/β-catenin signaling was required for differentiation of skeletal progenitors into osteoblastic lineage cells and also involved in the fibrous dysplasia, a disease that exhibits abnormal differentiation of skeletal progenitor cells. While Wnt/β-catenin signaling in osteoblast progenitors resulted in a fibrous dysplasia-like phenotype and reduction of β-catenin rescued differentiation defects of stromal cells. Activated Gα proteins, which act at β-catenin destruction complex assembly by binding Axin, played a physiological role during skeletal development and disease by modulating Wnt/β-catenin signaling strength [43]. Besides, Wnt/β-catenin signaling actively functioned in neurogenesis of hippocampus and midbrain [44,45], and Wnts like Wnt1 and Wnt3a promoted dopamine differentiation of neural progenitors and transplant of Wnts-primed neural stem cells showed improvements in model of Parkinson disease [46-49].
In this study, our data indicated that suitable working doses of TSA might be 50-500ng/ml for promotion of neural-linage differentiation of BM-MSC in vitro, while TSA doses less than 10ng/ml showed no significant effect, and doses higher than 500ng/ml showed more MSC toxicity. TSA is known as an anti-cancer agent for clinical usage, Wu et al found apoptosis-inducing effect by TSA at 50ng/ml in A549 cells, a non-small-cell lung cancer cell line. They demonstrated TSA resulted in apoptosis of A549 cells by increasing expression of TNFR and activation of caspase signaling pathway [50]. Besides, Liu et al also reported apoptosis of prostate cancer cells induced by treatment of TSA at concentration of 1mM [51]. Whether or not TSA exhibits similar effects on MSC in situ or transplanted MSC in vivo should need further investigation by animal experiment.
Finally, we especially focused on possible role of Wnt/β-catenin signaling in TSA effect on MSC in this study, and found that TSA-induced neural-linage commitment from BM-MSC might be possibly attained by triggering activation of Wnt/β-catenin signaling and modulation of down-stream gene expression regulation cell differentiation and proliferation. Western blot confirmed IWR1 blocking effect on Tuj-1, NG2, GFAP and CNPase expression of cultured MSC induced by TSA treatment. Data of present study supported our point that Wnt signaling might be possibly required for neural-linage cell differentiation of MSC. It is documented that Wnt/β-catenin signaling regulates cell adhesion, cell morphology, and gene expression controlling cell differentiation and proliferation [37,38], while HDAC inhibitor is also involved in gene transcription, cell proliferation, differentiation and apoptosis by modulating DNA activity. Result of this study further indicated that activation of Wnt/ β-catenin signaling induced by TSA treatment might be involved in regulation of cell division and neural-linage differentiation of BM-MSC [39,40]. Besides, the enhanced TNF receptor, increased somatostatin receptor, cyclooxygenase-2 activity, ERK signaling inhibition, BCL-2 down-regulation, increased in cell cycle G1 arrest, decreased in cyclin D1, D2 and cyclin dependent kinases, and activation of down-stream caspase might be involved in mediation of TSA effect [50-55]. In addition, HDAC inhibitor induced E-cadherin expression and membrane localization of E-cadherin/β-catenin complex, leading to reduced cancer cell migration and invasion [56]. HDAC inhibitor TSA was thus applied as an effective DNA damage strategy in human neuroblastoma or effective anti-cancer agent [50- 57]. It is hopefully that TSA-induced generation of neuronal cells may result in establishing new intervention for MSC transplant therapy that characterizes with lower tumorigenicity or higher safety.
In conclusion, TSA could induce both cell division arrest and neural-linage cell differentiation of cultured BM-MSC dose-dependently most possibly by triggering activation of Wnt/β-catenin signaling. These biological effects of TSA intervention indicated its beneficial roles in lowering tumorigenicity of transplanted MSC and increasing MSC differentiation efficiency into neurons or glial cells, strongly suggesting that TSA may be utilized as a potential candidate drug in manipulation of MSC therapy for treatment of various neurological disorders in human beings.
Acknowledgments
This word is supported by grants from National Basic Research Program of China (2011CB504103 and 2012CB525002) and National Natural Science Foundation of China (81272346, 81301036 and 31371374).
References
- Pevsner-Fischer M, Levin S, Zipori D. The origins of mesenchymal stromal cell heterogeneity. Stem Cell Rev. 2011; 7: 560-568.
- Satija NK, Gurudutta GU, Sharma S, Afrin F, Gupta P, et al. Mesenchymal stem cells: molecular targets for tissue engineering. Stem Cells Dev. 2007; 16: 7-23.
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006; 8: 315-317.
- Chen BY, Wang X, Chen LW, Luo ZJ. Molecular targeting regulation of proliferation and differentiation of the bone marrow-derived mesenchymal stem cells or mesenchymal stromal cells. Curr Drug Targets. 2012; 13: 561-571.
- Baqir S, Smith LC. Inhibitors of histone deacetylases and DNA methyltransferases alter imprinted gene regulation in embryonic stem cells. Cloning Stem Cells. 2006; 8: 200-213.
- Kim HJ, Rosenfeld MG. Epigenetic control of stem cell fate to neurons and glia. Arch Pharm Res. 2010; 33: 1467-1473.
- Ma DK, Marchetto MC, Guo JU, Ming GL, Gage FH, et al. Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat Neurosci. 2010; 13: 1338-1344.
- Sun J, Sun J, Ming GL, Song H. Epigenetic regulation of neurogenesis in the adult mammalian brain. Eur J Neurosci. 2011; 33: 1087-1093.
- Balasubramaniyan V, Boddeke E, Bakels R, Küst B, Kooistra S, et al. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience. 2006; 143: 939-951.
- Chen B, Dodge ME, Tang W, Lu J, Ma Z, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009; 5: 100-107.
- Hudson J, Titmarsh D, Hidalgo A, Wolvetang E, Cooper-White J. Primitive Cardiac Cells from Human Embryonic Stem Cells. Stem Cells Dev. 2012; 21: 1513-1523.
- Siebzehnrubl FA, Buslei R, Eyupoglu IY, Seufert S, Hahnen E, et al. Histone deacetylase inhibitors increase neuronal differentiation in adult forebrain precursor cells. Exp Brain Res. 2007; 176: 672-678.
- Covic M, Karaca E, Lie DC. Epigenetic regulation of neurogenesis in the adult hippocampus. Heredity. 2010; 105: 122-134.
- Kuzumaki N, Ikegami D, Tamura R, Sasaki T, Niikura K, et al. Hippocampal epigenetic modification at the doublecortin gene is involved in the impairment of neurogenesis with aging. Synapse. 2010; 64: 611-616.
- Kuzumaki N, Ikegami D, Tamura R, Hareyama N, Imai S, et al. Hippocampal epigenetic modification at the brain-derived neurotrophic factor gene induced by an enriched environment. Hippocampus. 2011; 21: 127-132.
- Yoo DY, Kim W, Nam SM, Kim DW, Chung JY, et al. Synergistic effects of sodium butyrate, a histone deacetylase inhibitor, on increase of neurogenesis Induced by pyridoxine and increase of neural proliferation in the mouse dentate gyrus. Neurochem Res. 2011; 59: 722-728.
- Lyssiotis CA, Walker J, Wu C, Kondo T, Schultz PG, et al. Inhibition of histone deacetylase activity induces developmental plasticity in oligodendrocyte precursor cells. Proc Natl Acad Sci USA. 2007; 104: 14982-14987.
- Saraiva NZ, Oliveira CS, Garcia JM. Histone acetylation and its role in embryonic stem cell differentiation. World J Stem Cells. 2010; 2: 121-126
- Wen S, Li H, Liu J. Epigenetic background of neuronal fate determination. Prog Neurobiol. 2009; 87: 98-117.
- Yao X, Zhang JR, Huang HR, Dai LC, Liu QJ, et al. Histone deacetylase inhibitor promotes differentiation of embryonic stem cells into neural cells in adherent monoculture. Chin Med J. 2010; 123: 734-738.
- Huang C, Xiang Y, Wang Y, Li X, Xu L, et al. Dual-specificity histone demethylase KIAA1718 (KDM7A) regulates neural differentiation through FGF4. Cell Res. 2010; 20: 154-165.
- Wu M, Zhang Y, Wu NH, Shen YF. Histone marks and chromatin remodelers on the regulation of neurogenin1 gene in RA induced neuronal differentiation of P19 cells. J Cell Biochem. 2009; 107: 264-271.
- Costa-Borges N, Santaló J, Ibáñez E. Comparison between the effects of valproic acid and trichostatin A on the in vitro development, blastocyst quality, and full-term development of mouse somatic cell nuclear transfer embryos. Cell Reprogram. 2010; 12: 437-446.
- Moschidou D, Mukherjee S, Blundell MP, Jones GN, Atala AJ, et al. Human mid-trimester amniotic fluid stem cells cultured under embryonic stem cell conditions with Valproic acid acquire pluripotent characteristics. Stem Cells Dev. 2012; 22: 444-458.
- Barnabé GF, Schwindt TT, Calcagnotto ME, Motta FL, Martinez G Jr, et al. Chemically-induced rat mesenchymal stem cells adopt molecular properties of neuronal-like cells but do not have basic neuronal functional properties. PLoS One. 2009; 4: e5222.
- Liu J, Lu XF, Wan L, Li YP, Li SF, et al. Suppression of human peripheral blood lymphocyte proliferation by immortalized mesenchymal stem cells derived from bone marrow of Banna Minipig inbred-line. Transplant Proc. 2004; 36: 3272-3275.
- Zurita M1, Bonilla C, Otero L, Aguayo C, Vaquero J. Neural transdifferentiation of bone marrow stromal cells obtained by chemical agents is a shorttime reversible phenomenon. Neurosci Res. 2008; 60: 275-280.
- Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009; 27: 1050-1056.
- Jeong JO, Han JW, Kim JM, Cho HJ, Park C, et al. Malignant tumor formation after transplantation of short-term cultured bone marrow mesenchymal stem cells in experimental myocardial infarction and diabetic neuropathy. Circ Res. 2011; 108: 1340-1347.
- Røsland GV, Svendsen A, Torsvik A, Sobala E, McCormack E, et al. Long-term cultures of bone marrow-derived human mesenchymal stem cells frequently undergo spontaneous malignant transformation. Cancer Res. 2009; 69: 5331-5339.
- Dressel R, Schindehütte J, Kuhlmann T, Elsner L, Novota P, et al. The tumorigenicity of mouse embryonic stem cells and in vitro differentiated neuronal cells is controlled by the recipients’ immune response. PLoS One. 2008; 3: e2622.
- Abumaree M, Al Jumah M, Pace RA, Kalionis B. Immunosuppressive Properties of Mesenchymal Stem Cells. Stem Cell Rev. 2012; 8: 375-392
- Nasef A, Ashammakhi N, Fouillard L. Immunomodulatory effect of mesenchymal stromal cells: possible mechanisms. Regen Med. 2008; 3: 531-546.
- Lei J, Wang Z, Hui D, Yu W, Zhou D, et al. Ligation of TLR2 and TLR4 on murine bone marrow-derived mesenchymal stem cells triggers differential effects on their immunosuppressive activity. Cell Immunol. 2011; 271: 147-156.
- Park MJ, Shin JS, Kim YH, Hong SH, Yang SH, et al. Murine mesenchymal stem cells suppress T lymphocyte activation through IL-2 receptor a (CD25) cleavage by producing matrix metalloproteinases. Stem Cell Rev. 2011; 7: 381-393.
- Yagi H, Soto-Gutierrez A, Parekkadan B, Kitagawa Y, Tompkins RG, et al. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2011; 19: 667-679.
- Widelitz R. Wnt signaling through canonical and non-canonical pathways: recent progress. Growth Factors. 2005; 23: 111-116.
- 38 Prakash N, Wurst W. A Wnt signal regulates stem cell fate and differentiation in vivo. Neurodegener Dis 2007; 4: 333-338.
- De Boer J, Wang HJ, Van Blitterswijk C. Effects of Wnt signaling on proliferation and differentiation of human mesenchymal stem cells. Tissue Eng. 2004; 10: 393-401.
- Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2009; 433: 1-7.
- Petrov N, Zhidkova O, Serikov V, Zenin V, Popov B. Induction of Wnt/β-catenin signaling in mouse mesenchymal stem cells is associated with activation of the p130 and E2f4 and formation of the p130/Gsk3β/β-catenin complex. Stem Cells Dev. 2012; 21: 589-597.
- Rodríguez-Carballo E, Ulsamer A, Susperregui AR, Manzanares-Céspedes C, Sánchez-García E, et al. Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways. J Bone Miner Res. 2011; 26: 718-729.
- Regard JB, Cherman N, Palmer D, Kuznetsov SA, Celi FS, et al. Wnt/β-catenin signaling is differentially regulated by Ga proteins and contributes to fibrous dysplasia. Proc Natl Acad Sci USA. 2011; 108: 20101-20106.
- Castelo-Branco G, Andersson ER, Minina E, Sousa KM, Ribeiro D, et al. Delayed dopaminergic neuron differentiation in Lrp6 mutant mice. Dev Dyn. 2010; 239: 211-221.
- Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005; 437: 1370-1375.
- Andersson ER, Prakash N, Cajanek L, Minina E, Bryja V, et al. Wnt5a regulates ventral midbrain morphogenesis and the development of A9-A10 dopaminergic cells in vivo. PLoS One. 2008; 3: e3517.
- Castelo-Branco G, Wagner J, Rodriguez FJ, Kele J, Sousa K, et al. Differential regulation of midbrain dopaminergic neuron development by Wnt-1, Wnt-3a, and Wnt-5a. Proc Natl Acad Sci USA. 2003; 100: 12747-12752.
- Parish CL, Castelo-Branco G, Rawal N, Tonnesen J, Sorensen AT, et al. Wnt5a-treated midbrain neural stem cells improve dopamine cell replacement therapy in parkinsonian mice. J Clin Invest. 2008; 118: 149-160.
- Tang M, Miyamoto Y, Huang EJ. Multiple roles of beta-catenin in controlling the neurogenic niche for midbrain dopamine neurons. Development. 2009; 136: 2027-2038.
- Wu TC, Yang YC, Huang PR, Wen YD, Yeh SL. Genistein enhances the effect of trichostatin A on inhibition of A549 cell growth by increasing expression of TNF receptor-1. Toxicol Appl Pharmacol. 2012; 262: 247-254.
- Liu Z, Marquez M, Nilsson S, Holmberg AR. Incubation with somatostatin, 5-aza decitabine and trichostatin up-regulates somatostatin receptor expression in prostate cancer cells. Oncol Rep. 2008; 20:151-154.
- Choi YH. Induction of apoptosis by trichostatin A, a histone deacetylase inhibitor, is associated with inhibition of cyclooxygenase-2 activity in human non-small cell lung cancer cells. Int J Oncol. 2005; 27: 473-479.
- Singh T, Prasad R, Katiyar SK. Inhibition of class I histone deacetylases in non-small cell lung cancer by honokiol leads to suppression of cancer cell growth and induction of cell death in vitro and in vivo. Epigenetics. 2013; 8: 54-65.
- Jasek E, Lis GJ, Jasinska M, Jurkowska H, Litwin JA. Effect of histone deacetylase inhibitors trichostatin A and valproic acid on etoposide-induced apoptosis in leukemia cells. Anticancer Res. 2012; 32: 2791-2999.
- Yao J, Qian CJ, Ye B, Zhang X, Liang Y. ERK inhibition enhances TSA-induced gastric cancer cell apoptosis via NF-?B-dependent and Notch-independent mechanism. Life Sci. 2012; 91: 186-193.
- Catalano MG, Fortunati N, Pugliese M, Marano F, Ortoleva L, et al. Histone deacetylase inhibition modulates E-cadherin expression and suppresses migration and invasion of anaplastic thyroid cancer cells. J Clin Endocrinol Metab. 2012; 97: E1150-1159.
- Poljakova J, Hrebackova J, Dvorakova M, Moserova M, Eckschlager T,, et al. Anticancer agent ellipticine combined with histone deacetylase inhibitors, valproic acid and trichostatin A, is an effective DNA damage strategy in human neuroblastoma. NeuroEndocrinol Lett. 2011; 32 Suppl 1: 101-116.