
Research Article
J Stem Cell Res Transplant. 2014;1(2): 1012.
Neurogenic Stem Cells Have the Capacity to Disperse Widely and Fuse with Host Neurons in Adult Rats
Emily Kimes, Michele Kanemori, Daniella Amri, Ashley Noone, Jerika Barron, Ashley Saito, Omar Cortez-Toledo, Ellie Cortez-Toledo, David Arrizon, Johanna Quist, Yohualli Balderas, Melissa Miranda, Tina Tran, Frances Kim, and Kerry Thompson*
Occidental College, VA Greater Los Angeles Healthcare Center, University of California, Los Angeles, USA
*Corresponding author: Kerry Thompson Ph.D, Occidental College, VA Greater Los Angeles Healthcare Center, University of California, Los Angeles, USA
Received: September 01, 2014; Accepted: October 17, 2014; Published: October 20, 2014
Keywords
Neural stem cells; Fusion; Transplantation; Cortical pyramidal cells; Purkinje cells
Introduction
The central nervous system (CNS) has a relatively limited capacity for neurogenesis, and self-repair. As a result, diseases that produce neural losses are particularly devastating. The greater the amount of neural tissue loss, the greater the impact on the functioning of the patient. Traumatic brain injury and degenerative CNS diseases such as Alzheimer's disease, Parkinson's disease, and intractable temporal lobe epilepsy (TLE), are clinical challenges that could benefit from the development of cell replacement therapies. For these patient populations, the ability to replace lost brain tissue with functional neurons and glia, is an ambitious, but worthwhile goal. Embryonic stem cells (ESC) that can be genetically engineered, propagated and differentiated, or "neuralized", for subsequent transplantation into the central nervous system, would offer a means to accomplish this important goal [1-5].
Rapid progress is being made in the field of ESC-derived neuron generation and characterization [6]. In most studies, ESC are exposed to a series of chemical treatments that push their differentiation down a neural pathway that can be characterized biochemically. Many related studies have demonstrated proof-of-principal for the transplantation of these cell lines into animal model systems [7-9]. It has been reported that neuralized ESC that have been genetically engineered with reporter molecules, remain viable in the host for long periods [10] and, remarkably, that fully differentiated neural phenotypes expressing transgenes can be found in the host within 30d of transplantation [11]. These findings are important milestones toward the development of neuron replacement therapy, but the interpretation of these studies is challenged by recent reports of the capacity for ESC-derived neurons to fuse with host neurons.
The concept of stem cell fusion in the brain is not new to the literature. In fact, early reports of what was initially believed to be bone marrow derived stem cell (BMDC) trans differentiation in the cerebellum [12] was later discovered to be either complete [13-15] or partial [16] fusion of BMDC with fusionogenic cerebellar Purkinje cells. In the absence of tissue damage BMDC fusion occurs only rarely, and it is not found in brain cells outside of the cerebellum. Presumably because these are rare events, and seemingly specific to BMDC and Purkinje cells, the phenomena has not been consistently pursued as a potential contributor to the histological outcomes following ESCderived neural cell transplantation. Recently, however, studies have shown that fusion does, in fact, occur following transplantation of ESC-derived neural cells and, similar to the BMDC studies, particular cell types seem to be more fusionogenic [11-17].
We have been pursuing transplantation strategies for the treatment of intractable neurological diseases like TLE using genetically modified neural cells [18-22]. In our previous studies using neural cell lines of non-stem cell origin, we have never seen evidence of widespread dispersion or fusion [20-23]. We report here that genetically modified neurogenic stem cells have the capacity to disperse widely following cerebral transplantation into the striatum, and to fuse with host neurons. As has been recently reported [11-13], cortical pyramidal cells are highly fusionogenic, but we believe the fusion-competent cells are not limited to only this cell type. These data are consistent with transplantation results extending back many years and may call for a re-interpretation of those data to include potential fusion events.
Materials and Methods
Cells and cell culture
The feeder free ZHTc6 cell line (24) was purchased from the Institute for Stem Cell Research (Edinburgh, UK). This line was created by genetically engineering wild-type CGR8 murine embryonic stem cells to constituitively express a transactivator molecule (tTA), and one allele of the Pou5f1 gene (which codes for Oct-3/4) under the transcriptional control of a tTA-sensitive promoter (hCMV*- 1). The Oct-3/4 expressing construct is bicistronic and also codes for the reporter molecule beta-galactosidase (β-gal) fused to a neomycin resistance gene driven by the same promoter. In this cell line, up-regulation of OCT-3/4 expression (in the absence of tetracycline), combined with the removal of serum, and leukemia inhibitory factor (LIF), has neurogenic effects [25]. The cells were cultured on gelatinized plates in media containing the following: Glasgow Minimum Essential Medium (Sigma), 2 mM glutamine, 1 mM sodium pyruvate (Gibco), nonessential amino acids (Gibco), 10-15% (v/v) fetal bovine solution (Invitrogen), 1:1000 dilution of β-mercaptoethanol (Sigma), and 500-1000 units per ml of LIF, (Millipore) and grown on 0.1% gelatinized plates and then incubated at 37.0 C and 7.0% CO2 until islands formed. Media was replaced daily. In vitro differentiation was performed by eliminating LIF and growing the cells in media with 4% serum (modeling transplantation into the host).
In vitro histochemistry and immunocytochemistry
To stain stem cell cultures, cells were grown to 70-90% confluence in 6-well dishes and then washed and fixed in a 0.25% glutaraldehyde solution for 15-30 min. The fixative was removed; the cells were washed thoroughly and then incubated at 37.0oC in 1ml of an X-gal solution prepared according to the manufacturer's recommendations (Promega Corporation). The solution was removed 1-18 hrs later, and then the cells were washed and analyzed using an inverted scope and regular light microscopy. In some differentiation experiments, cells were fixed, permeabilized, and incubated with an anti-Nestin antibody (Chemicon International) followed by exposure to a fluorescein-conjugated secondary antibody.
Animals
Sprague Dawley females were purchased from Taconic Biosciences Inc. (Hudson, NY). Animals were 200-250 grams (approximately 2-3 mo (N=35)). Rats were housed under a 12-h light/ dark cycle with free access to food and water. All procedures were performed in accordance with the IACUC guidelines approved by Occidental College.
Unilateral 6-hydroxydopamine (6-OHDA) lesion
In a subset of animals, dopaminergic neurons were unilaterally targeted by focal administration of 6-OHDA into the medial forebrain bundle (MFB). Rats were anesthetized with a mixture of xylazine: ketamine (10mg/kg:85mg/kg) or with isoflurane vapor using a vaporizer. Rats were injected with 25 mg/kg (i.p.) desipramine, 30 minutes before 6-OHDA injection, in order to prevent uptake of 6-OHDA by noradrenergic neurons. Stereotaxic surgery was performed to deliver 6-OHDA unilaterally at two locations within the MFB: 2.5μl (0.5μl/2min) AP -4.4, ML -1.2, DV -7.8, and 2.0 μl (0.5 μl/2min) AP -4.0, ML -0.8, DV -8.0, relative to bregma [26-28]. 6-OHDA was administered as 3 μg/μl in a 1% ascorbic acid solution. To minimize oxidation, 6-OHDA solutions were freshly made, kept on ice, and light-protected. Animals were kept on a 37.0?C warming pad during surgery and in a 37.00C recovery chamber after the surgery. A solution of 5% dextrose was injected subcutneously after surgery at 10% body weight.
Five to six days after surgery rats were behaviorally screened for the extent of the 6-OHDA lesion using the amphetamine rotation test. Rats were placed in a Rotomax rotometer (AccuScan Instruments, Inc., Columbus OH), which consists of a 12" diameter Plexiglas cylinder on a flat surface. A harness extends down and permits the animals to touch the wall of the cylinder. Animals were injected with 5 mg/kg amphetamine sulfate and the number turns/5 minutes were calculated from net ipsilateral turns over 30 minutes following a five minute habituation period. Peak responses were anticipated within this time period [29]. Rats that rotated at least 5 ipsilateral (to lesion) turns per minute (net) were designated as "positive responders" and animals that rotated below that level were designated "negative responders". The negative responders were used as an additional "non-lesioned"control group. Animals were returned to their home cages for a period of 2-4 weeks before they received stem cell transplantation into the striatum that was targeted for denervation.
Transplantation
On the day of surgery, animals were injected with a combination of xylazine and ketamine or exposed to isoflurane vapor to produce surgical anesthesia. Once the animals were placed into the stereotaxic apparatus they were injected with neurogenic ESCs unilaterally into the striatum. The majority of the animals were injected with undifferentiated cells to promote cell survival after transplantation. Three of animals were transplanted with cells that were grown under differentiating conditions for three days. The cells were washed and suspended in sterile PBS at 150K cells/μl and held on ice for no longer than two hours. Three injections were made into the striatum at the following coordinates relative to bregma: AP 0.0, ML -1.2, and DV -5.5, 4.5 3.5 with 1.0 μl delivered over a two minute period to each site. Prior to injection of ESC, the surgery needle was washed with ethanol followed by thorough PBS washes. Animals were kept on a 37.00C warming pad during surgery and then placed in a 37.00C recovery chamber after the surgery until they regained their righting reflex. The surgical controls that were used for comparison were injections of dead cells (exposure to hypertonic conditions, N=4), predifferentiated cells (LIF removal and serum reduction, N=3) and current and historical PBS controls (N=1 and N=4 respectively
Histology
At multiple pre-determined survival times after the transplantation of cells, the animals were transcardially perfused with cold 4% paraformaldehyde or a 2% paraformaldehyde and .5% glutaraldehyde mixture (for EM processing) and the brains (and liver samples were removed and cryoprotected in 30% sucrose. Brains were recovered 24hr (N=3), 3 days (N=3), 7days (N=3), 2 wks (N=3), and 4wks (N=18 (13) lesioned and (5) unlesioned). The tissue was serially sectioned using a cryostat by blocking and cutting the whole cerebrum (coronal plane), cerebellum (sagittal plane), and liver samples by taking slices at 20, 40, or 60 depending on the histological protocol. Adjacent sections were used for hematoxylin and eosin (H&E) staining and X-gal histochemistry (each 20 μ) and were typically taken every 200μ with adjacent 40μ or 60μ sections taken for immunohistochemistry or TEM, respectively. Tissue was sampled more frequently in the plane of the cell injection to maximize observations in the region of highest interest, which was the transplanted striatum and the surrounding cortex. For H&E staining and X-gal histochemistry, sections were cut onto glass slides. For immunohistochemistry and TEM, sections were placed into PBS.
X-gal Histochemistry
β-gal positivity was used to analyze and locate the β-gal-positive cells in the brain and liver tissue sections. Glass-mounted sections of both brain and liver, and 60μ free-floating sections of the brain, were pretreated by permeabilization with 0.01% deoxycholic acid and 0.02% Igepal in PBS at RT. The slides were then thoroughly washed and the mounted sections were encircled using a PAP pen or grease, so that each of the sections could be incubated in a filtered X-gal solution containing 5 mM potassium ferrocyanide, 5mM potassium ferricyanide, 2mM MgCl2, and 250 μg/ml X-gal in PBS (pH 7.4 or 8.5 to discriminate bacterial β-gal from endogenous enzyme sources) within a dark box placed within a humidified chamber and 37oC. The sections were left to incubate overnight. Sections were then rinsed, mounted onto glass slides if necessary and cover slipped the next morning. Tissue sections were left to dry and then evaluated with conventional light microscopy. Negative controls were incubated for equivalent periods.
Quantification
X-gal stained tissue was used to quantify areas and numbers of β-gal-positive cells. The tissue section with the largest representation of the injection track, from each of the lesioned-transplanted animals, was used to assess the cross-sectional area of the transplants in the injected striatum. The sections were viewed using a 4X objective and digital images were taken. The ImageJ program was used to outline the perimeter of the transplant and then to calculate the area (N=3 for 1, 3, 7, and 14d, and N=5 for 1 month). Additionally, in the one month post transplant animals, the number of β-gal-positive Purkinje cells was counted in lobes III, IV, and V of the cerebellum [30]. Comparable 1mm sections were targeted in the selected lobes and the total number of β-gal-positive cells were compared to the total number of Purkinje cells in those regions.
Immunohistochemistry
Indirect immunofluorescence and immunoperoxidase histochemical strategies were performed on free-floating sections. A polyclonal anti β -gal antibody (Chemicon International) was used at [1:1000], an anti-GFAP antibody (Millipore) was used at [1:1200] and an anti-NeuN (Millipore) antibody was used at [1:500]. Speciesappropriate secondary antibodies were used with either fluorescein or Texas red fluorescence (Vector Labs). For double labeling experiments, antibodies were exposed serially to the tissue sections. In some cases, following the secondary antibody exposure, the sections were exposed to Hoechst (1:1000) to fluorescently label all nuclei. For immunoperoxidase staining, an ABC kit (Vector Labs) was used in accordance with the manufacturer's instructions. In all cases, tissue sections were incubated in the primary antibodies overnight at 4°C.
Immunohistochemical controls included the omission of the primary antibodies, the secondary antibodies, and, in some cases, the inclusion of an endogenous biotin-blocking procedure (Vector Labs). All of these sections were examined by conventional light and fluorescent microscopy and a subset of the tissues were evaluated using a confocal microscope (Leica TCS-SP2 AOBS inverted confocal Microscope (UCLA core facility).
Transmission electron microscopy
A modification of a previously published procedure combining X-gal histochemistry and TEM [16] was used. For a subset of animals at the 30 day survival time point, frozen sections were cut at 60 and then the free-floating sections were reacted with the X-gal solution overnight. The reacted tissue was mounted onto glass slides and then viewed under a dissecting microscope. Cortical sections near the plane of injection, with identified X-gal-positive cells, were excised with a razor blade and then processed for TEM (5-7 excisions per animal). The excised tissue was rinsed for 10 min in 0.1M Na cacodylate buffer and then in 1% osmium made in the same buffer, for 2 hrs. After a second short rinse, the tissue was dehydrated in graded alcohols and then infiltrated with Spurs overnight. The next day the tissue was placed into molds and then baked for at least 24 hrs. Thick sections (1.5) were cut from blocks using a LKB Nova ultramicrotome, they were then counterstained with toludine, and viewed under a light microscope. Blocks producing thick sections that contained cells with evidence of X-gal precipitate and/or with profiles that were consistent with heterokaryon, were selected for further analysis by obtaining thin sections (70nm) that were evaluated using a Zeiss EM 109 transmission electron microscope.
Results
We confirmed that the ZHTc6 line expresses Oct 4 and SSEA-1 (a marker associated with neural stem cell precursors), in the absence of doxycycline (data not shown). In proliferation media, the cells did retain undifferentiated phenotypes, but a fraction would flatten, and many of those cells extended processes. In differentiation media the majority of the cells underwent changes in morphology with stereotypic neuritic projections. Cell division halted after ~3 days in differentiation media and a significant percentage of cells died over a period of five days. The neuritic projections of the adherent differentiated cells, at five days, were positive for the CNS progenitor marker nestin (Figure 1E). The ZHTc6 cells and all of their derivatives, express β-galactosidase in the absence of doxycycline (Figure 1B). This made visualization of the transplanted cells possible by: 1) exposing histological sections to the substrate X-gal which produces a blue reaction product only within the enzyme-containing cells and 2) performing immunohistochemistry using anti-β-galactosidase antibodies. We used both of these techniques simultaneously in all animals. We found that the overall patterns of cellular staining were the same, but that immunohistochemistry was more sensitive as it revealed more cells and their processes (Figures 3 and 4). We also performed nissl staining on adjacent sections for every animal.
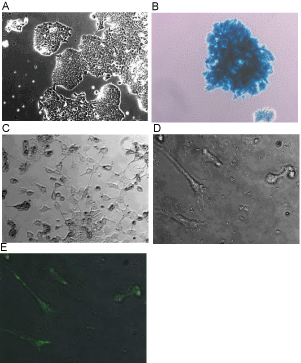
Figure 1: ZHTc6 mouse embryonic stem cells express β-galactosidase and
are neurogenic. A) When grown in high serum and LIF containing media
the cell lines show growth characteristics typical of ESC lines. B) X-gal
histochemistry shows that all of the cells carry the reporter molecule. C)
When the serum concentrations are lowered and LIF is omitted from the
media over five days, the cells differentiate and extend neurites. D and E)
Anti-Nestin antibodies labeled cells that had neural phenotypes in culture
(cells were grown in differentiation media for 5 days. D is a bright field image
and E is an overlay showing secondary antibody fluorescence). Images were
taken with the following objectives: 10X (A&B), 20X (C), and 40 X (D&E).
Regardless of the detection method, the time course data showed that a large bolus of cells could be found in the target sites within the striatum and along the needle track (Figure 2). The data suggest that when undifferentiated neurogenic stem cells are transplanted, they continue to divide for a matter of days or weeks in a non-linear manner. We measured the cross-sectional area of the transplants in tissue sections with the largest representation of the transplanted cells (N=3 at early time points, N=5 at 30 days) and found that the borders of the transplanted region widened out to three days, roughly plateaued between three and seven days, and then underwent a second round of expansion (suggesting expansion of surviving undifferentiated cells) (Figure D). At 24 hrs the area (in mm2) was .53 ± .13, at 3d it was 1.3 ± .64, at 7d it was 2.28 ± .82, and at 14d it was 7.32 ± 1.5. By one month however, the transplant core within the striatum dramatically contracted leaving relatively small tracks and few cells compared to the 2 wk time point (05 ± .02. mm2) (Figure 2E). As early as three days, and increasing in number by seven days, cells at the margins were seen separating from the core of the transplant (Figure 2B inset) as that margin became increasingly irregular. By two weeks, the borders of the core became irregular as the cells either continue to divide, differentiate, or die off in numbers.
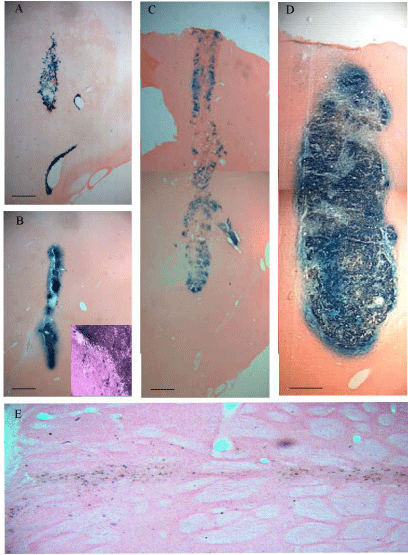
Figure 2: Growth and survival of transplanted neurogenic stem cells over
time. Sections recovered from the plane of the injection needle track show
that cells continue to express β-gal in the target site at A) 24 hr B) 3d C) 7d
D) 14 d and E) one month following transplantation into the striatum. As early
as 3d the cells were observed outside the margins of the transplant. B inset)
The dark staining shows the core of the transplant and transplanted cells
can be seen escaping those margins at higher magnification. E) By 30d the
core of the transplant was greatly contracted relative to the 14d time-point
but β-gal-positive cells with neuronal profiles persisted. The scale bars for
A-D are 500μ. The scale bar for E is 50μ. Figure 2B (inset) was taken with a
40X objective.
Outside the core the presence of β-gal-positive cells was inversely related to number of cells within the core. Staining became increasingly prominent in non-targeted areas distal to the transplanted striatum beginning at one week following transplantation and, by two weeks, the distribution of cells was well established bilaterally in predictable regions (vasculature, thalamus and midbrain, pyramidal cells of the cortex, and in the cerebellum (Purkinje cells and deep cerebellar nuclei)) (Figure 5). When blood vessels were cut in cross section, cellular staining was present in the vessels, suggesting intravasation. There was also evidence of extravasation because the liver showed islands of positively stained cells (Figure 6).
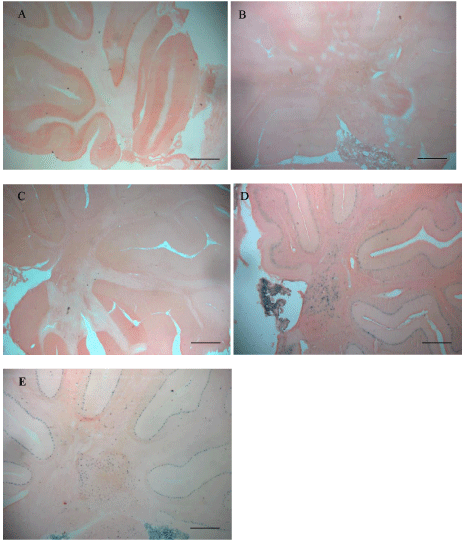
Figure 5: The prevalence of β-gal-positive cells increased over time in
regions distal from the striatal injection site. One day (A), 3d (B), and 7d (C),
positively stained cells in the cerebellum were much less frequent than at 14d
(D) and 30d (E). By the later time points cells were distributed throughout
the brain but were especially prominent in the Purkinje cells, deep cerebellar
nuclei, and the choroid plexus (D). Scale bars set at 500μ.
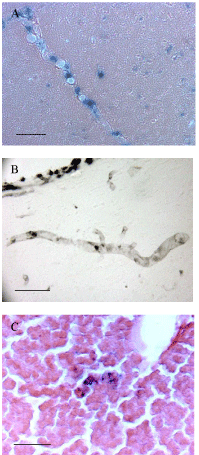
Figure 6: Transplanted cells gained access to the vasculature of the host
and showed the capacity to intravasate and extravasate. A and B) When
histological sections exposed blood vessels in cross section, stem cells were
revealed within those vessels using A) X-gal histochemistry or B) indirect
immunohistochemistry. C) To investigate whether the cells were entering
other tissues through the general circulation liver samples were evaluated.
Islands of positive cells could be detected in these tissues (C). Scale bars
are set at 50μ.
By 30 days after transplantation, a large number of positivelystained mature cortical pyramidal cells were evident regardless of the staining technique. These β-gal cells were commonly found in the cortex overlaying the striatum. Immunoperoxidase and immunofluoresecent images produced profiles of discrete pyramidal cells and their processes extending throughout these cortical sections (Figure 3). Interestingly, fluorescent nuclear staining often demonstrated small condensed nuclei opposed to the large, dispersed nuclei of the pyramidal cells, (Figure 3C and 3D). In the cerebellum too, multiple methods revealed large Purkinje cells with their distinctive dendritic trees stained positively for the stem cells marker β-gal (Figure 4B). The β-gal-positive cells co-labeled with the neuronal marker NeuN but not the glial marker GFAP (Figure 7). When representative areas of the Purkinje cell layer were evaluated for the presence of X-gal positive cells at the 30 day time point, as many as 85% of the cells showed evidence of staining (432 positive cells out of 505 cells sampled).
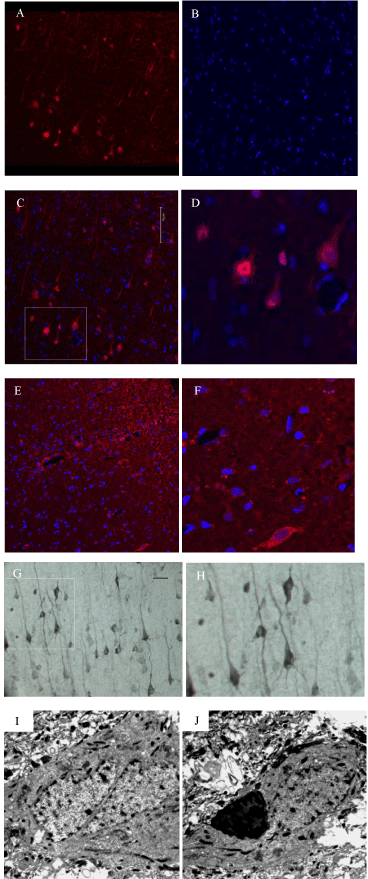
Figure 3: Fully mature cellular profiles in the cortex and striatum stain
positively for the stem cell marker β-gal and some show evidence of
binucleation. A-F) Immunofluorescence detecting the stem cell marker β-gal,
in transplanted tissue, shows mature cortical pyramidal neurons and their
processes (A-D) and cells in the striatum adjacent to the needle track (E and
F). When the tissue is counter-stained with Hoechst (B-F), immunolabelled
labeled cells reveal large dispersed nuclei and, often, small condensed
satellites. D&F are enlargements of the outlined areas in C and E. G and
H) Immunoperoxidase staining for β-gal shows the fine neuritic processes
of the positively-labelled cortical pyramidal cells in the cortex overlaying the
transplanted striatum that have a laminar distribution (H is an enlargement
of the outlined area in G). I and J) TEM analysis of X-gal-positive cells
retrieved from the same areas shown in A-H shows that binucleation can
occur in these cells but, more commonly, β-gal-positive cells have condensed
electron dense profiles impacting their cell soma (J).The scale bar shown in
3C applies to micrographs A-F. Scale bar in G set at 50μ. TEM images are
shown at 4400X magnification.
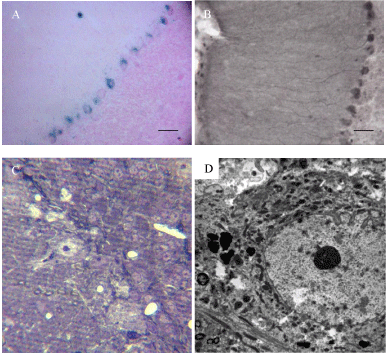
Figure 4: Cerebellar Purkinje cells stained positively for the reporter
molecule but did not show binucleation. A) X-gal reactions using reagents
that discriminate bacterial from endogenous reactions and B) indirect
immunohistochemistry with or without suppression of endogenous biotin,
produced clear Purkinje cell profiles. C) Thick (1.5 μ ) sections were used to
identify cells carrying the reaction product for analysis of ultrastructure (D).
No evidence of binucleation was found in the Purkinje cells. Scale bar set at
50μ for A and B, and 10μ for C.TEM image is shown at 3000X magnification.
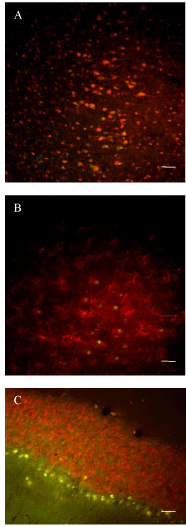
Figure 7: Transplanted cells positive for β-galactosidase co-express a
neuronal marker but not a glial marker. A-C) One month after transplantation
tissue sections were exposed to two antibodies serially to determine the
biochemical phenotype of the differentiated stem cells. A) In cortical sections
a polyclonal rabbit anti-β-gal antibody was used in combination with a mouse
monoclonal anti-NeuN antibody. The secondary anti-rabbit antibody was
conjugated to fluoroscein (green) while the secondary anti-mouse antibody
was conjugated to biotin and then exposed to an avidin-conjugated Texas
red molecule. There was considerable overlap in the two staining patterns
(colocalization is indicated by yellow/orange hues. B) Adjacent tissue was
exposed to a mouse monoclonal anti-β-gal antibody in combination with a
rabbit anti-GFAP antibody (B). In this case the secondary anti-mouse antibody
was conjugated to fluoroscein and the secondary anti-rabbit antibody was
biotinylated for subsequent visualization using an avidin-linked Texas red
molecule. There was no evidence of cells expressing both β-gal and GFAP
molecules in this case. C) The same strategy was applied to cerebellar tissue.
Texas red fluorescence indicated NeuN positivity and green fluorescence
indicated β-gal positivity. There was considerable overlap in the Purkinje cell
layer but none in the granule cell layer. Scale bars set at 50μ.
To investigate whether the β-gal-positive cells were binucleated, we combined histochemistry and TEM to study the ultrastructure of the largest of the positively staining cells by targeting the cortex overlaying the striatum and the Purkinje cell layer of the cerebellum. The β-gal reaction product was first identified in 60μ sections using light microscopy and then targeted tissue was excised and processed for viewing with a transmission electron microscope. Lightly counterstained thick sections (1.5 revealed a number of cortical pyramidal cells, and Purkinje cells that contained the histochemical reaction product. In thin sections the same cells were darkened by the reaction product and often contained granular electron-dense particles as has been previously reported [16]. We found evidence of clear binucleation in the cortex of two of three animals investigated (Figure 3I). Additionally, all of the animals showed β-gal-positive cortical and Purkinje cells opposed to highly condensed cells containing electron dense material (Figure 3J). In each animal we also found β-gal-positive cells within the cortex and midbrain that had no evidence of an additional associated cellular profile at the level of the cell body. This was also evidenced in the cerebellum where we examined over 100 cells that were positive for β-gal, but never showed binucleation and, only occasionally, showed a closely apposed β-galpositive cell (Figure 4C,D).
Discussion
We transplanted neurogenic stem cells into a commonly used rat model of Parkinson's disease and found evidence of widespread dispersion from the transplanted region in both lesioned and nonlesioned animals, and the capacity for transplanted cells to fuse with host neurons. The dispersion and fusion of the cells had initiated by the end of the first week following transplantation and it was fully established by the end of the second week when cell numbers were maximal at the transplant site. Despite contraction of the number of cells within the target area, the extra-striatal staining persisted in predictable cell populations out to one month. The areas of prominent staining for the stem cell-derived reporter molecule included, but were not restricted to, large cortical pyramidal cells above the targeted stiatum, and large Purkinje cells of the cerebellum. Both of these populations have been identified as fusionogenic in recent reports [11,16,17,31,32]. In the cortex there was ultrastructural evidence of binucleation in pyramidal shaped neurons. However, the majority of positively-staining cells did not show strict binucleation, although many were in close opposition to condensed cellular elements. These data suggest that both complete, and partial, cell fusion may occur after transplantation of neural cells derived from embryonic stem cells.
Cell fusion with binucleation has recently been shown to occur following transplantation of NSC into young [11] and adult animals [17]. These reports did not involve experimental lesions, or treatments other than stem cell neuralization, to promote the fusion events. The stem cell line used in the present study has been genetically engineered to over-express OCT ¾ which promotes a neuroectodermal fate [25]. We transplanted this clonal cell line into a commonly used model of 6-OHDA pretreatment. We were able to compare the fusion rates between lesioned and non-lesioned animals, in order to evaluate whether pathotropism could participate in the effect by looking at the lesioned vs. non-lesioned side. While cells expressing the transgene could be found in the target region of the denervated striatum (Figure 3E and 3F), they were not exclusively, or even predominantly, found in the lesioned substantia nigra, or in the denervated striatum, after one month in this hemiparkinson model. In fact, there was no difference in the dispersion pattern between lesioned and non-lesioned animals. The staining was found well away from the transplant site and in cell populations that are not believed to be affected by 6-OHDA pretreatment (e.g. Purkinje cells).
An obvious question is how the cells disperse so widely throughout the brain. Dead cells did not disperse and pre-differentiation of the cells limited dispersion, particularly into the cerebellum, but staining persisted outside the transplanted region even with that strategy. One potential explanation is that the cells were able to gain entry into the vasculature. Supporting this idea, when histological sections included large vessels in cross section, undifferentiated stem cell profiles were often found single file within those vessels. This was evident in both the X-gal staining and the β-gal immunohistochemistry (Figure 6A and 6B). Additionally, cells were frequently found within the choroid plexus which is a point of plasma filtration that produces cerebral spinal fluid (Figure 5D and 5E). To verify that the cells were entering the general circulation we also analyzed liver sections from the transplanted animals. Indeed, we occasionally found small islands of β-gal-expressing stem cells in the liver (Figure 6C). It seems that these neurogenic stem cells not only have the capacity for intravasation but also for subsequent extravasation. Fusion of BMDC with Purkinje cells is thought to occur following extravasation events and extravasation into the brain has been reported after intravenous injection of NSC as well [33].
An obvious question is how the cells disperse so widely throughout the brain. Dead cells did not disperse and pre-differentiation of the cells limited dispersion, particularly into the cerebellum, but staining persisted outside the transplanted region even with that strategy. One potential explanation is that the cells were able to gain entry into the vasculature. Supporting this idea, when histological sections included large vessels in cross section, undifferentiated stem cell profiles were often found single file within those vessels. This was evident in both the X-gal staining and the β-gal immunohistochemistry (Figure 6A and 6B). Additionally, cells were frequently found within the choroid plexus which is a point of plasma filtration that produces cerebral spinal fluid (Figure 5D and 5E). To verify that the cells were entering the general circulation we also analyzed liver sections from the transplanted animals. Indeed, we occasionally found small islands of β-gal-expressing stem cells in the liver (Figure 6C). It seems that these neurogenic stem cells not only have the capacity for intravasation but also for subsequent extravasation. Fusion of BMDC with Purkinje cells is thought to occur following extravasation events and extravasation into the brain has been reported after intravenous injection of NSC as well [33].
Another important question is what physiological phenomena drive the examples of neural stem cell fusion demonstrated here and in previous reports [11,17]. Physiological cell fusion occurs with high specificity between cells of the same lineage and can generate syncytium of trophoblasts and muscle cells. The fusion that leads to syncytia requires, among other things, phosphatidylserine externalization which follows reversible caspase 8 activation. Macrophages, inflammation-related cytokines, and a large number of other molecules are thought to participate in the processes that lead to chemotaxis, pore-formation, and reprogramming [34,35]. All of these elements likely participate in heterologous NSC fusion with host neurons subsequent to transplantation. If carried to completion, this leads to complete binucleation, but if events progress only to the formation of continuous pores and then halts, then partial cell fusion would result in transient sharing of cytoplasmic contents, including reporter molecules. The type of pore formation that could lead to partial cell fusion has been demonstrated using mesenchymal stem cells In vitro [36] and, in vivo, partial cell fusion of BMDC with cerebellar Purkinje cells has been shown to occur [16]. Transient cell fusion would help explain our findings in the Purkinje cells, and elsewhere, where the reporter molecule was present but binucleation was absent. These hypotheses need be thoroughly pursued beyond this descriptive study to make conclusive statements.
Here we have shown that stem cells that have been genetically modified to be neurogenic, and to express a reporter molecule, have the capacity to disperse widely in the host and undergo fusion with binucleation, and likely to a larger extent, partial fusion events. Widespread dispersion is likely mediated by access to the general circulation but the variables that permit neuron-specific fusion are unknown. Since there are overlapping physiological processes in physiological cell fusion and in the apoptotic cell death, and immunological responses that occur following transplantation into the developing and mature brain, one field may inform the other. What is clear is that close scrutiny must be applied to the histological outcomes following transplantation of ESC-derived neurons so that the contributions of complete and/or partial cell fusion are not overlooked. Additionally, a better understanding of fusion might be capitalized on as a strategy for enzyme replacement therapy in the brain [37].
These studies were supported by Grants from: The VA Merit Review Program, The Parkinson's Disease Research, Education, and Clinical Center, and The Occidental College Undergraduate Research Center.
The authors would like to thank Dr. Gary Martin for sharing resources and expertise in the experiments using electron microscopy.
References
- Aleynik A, Gernavage KM, Mourad Y, Sherman LS, Liu K, et al. Stem cell delivery of therapies for brain disorders. Clin Transl Med. 2014; 3: 24.
- Fargen KM, Mocco J, Hoh BL. Can we rebuild the human brain? The exciting promise and early evidence that stem cells may provide a real clinical cure for stroke in humans. World Neurosurg. 2013; 80: e69-72.
- Kim SU, Lee HJ, Kim YB. Neural stem cell-based treatment for neurodegenerative diseases. Neuropathology. 2013; 33: 491-504.
- Martinez-Morales PL, Revilla A, Ocana I, Gonzalez C, Sainz P, et al. Progress in stem cell therapy for major human neurological disorders. Stem Cell Rev. 2013; 9: 685-699.
- Yoo J, Kim HS, Hwang DY. Stem cells as promising therapeutic options for neurological disorders. J Cell Biochem. 2013; 114: 743-753.
- Srivastava AS, Malhotra R, Sharp J, Berggren T. Potentials of ES cell therapy in neurodegenerative diseases. Curr Pharm Des. 2008; 14: 3873-3879.
- Espuny-Camacho I, Michelsen KA, Gall D, Linaro D, Hasche A, et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron. 2013; 77: 440-456.
- Ideguchi M, Palmer TD, Recht LD, Weimann JM. Murine embryonic stem cell-derived pyramidal neurons integrate into the cerebral cortex and appropriately project axons to subcortical targets. J Neurosci. 2010; 30: 894-904.
- Neuser F, Polack M, Annaheim C, Tucker KL, Korte M. Region-specific integration of embryonic stem cell-derived neuronal precursors into a pre-existing neuronal circuit. PLoS One. 2013; 8: e66497.
- Nasonkin I, Mahairaki V, Xu L, Hatfield G, Cummings BJ, et al. Long-term, stable differentiation of human embryonic stem cell-derived neural precursors grafted into the adult mammalian neostriatum. Stem Cells. 2009; 27: 2414-2426.
- Cusulin C, Monni E, Ahlenius H, Wood J, Brune JC, et al. Embryonic stem cell-derived neural stem cells fuse with microglia and mature neurons. Stem Cells. 2012; 30: 2657-2671.
- Crain BJ, Tran SD, Mezey E. Transplanted human bone marrow cells generate new brain cells. J Neurol Sci. 2005; 233: 121-123.
- Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, et al. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003; 425: 968-973.
- Kozorovitskiy Y, Gould E. Stem cell fusion in the brain. Nat Cell Bio. l2003; 5: 952-954.
- Weimann JM, Johansson CB, Trejo A, Blau HM. Stable reprogrammed heterokaryons form spontaneously in Purkinje neurons after bone marrow transplant. Nat Cell Biol. 2003; 5: 959-966.
- Nern C, Wolff I, Macas J, von Randow J, Scharenberg C, et al. Fusion of hematopoietic cells with Purkinje neurons does not lead to stable heterokaryon formation under noninvasive conditions. J Neurosci. 2009; 29: 3799-3807.
- Brilli E, Reitano E, Conti L, Conforti P, Gulino R, et al. Neural stem cells engrafted in the adult brain fuse with endogenous neurons. Stem Cells Dev. 2013; 22: 538-547.
- Anantharam V, Behrstock S, Thompson K, Weatherwax R, Bongarzone E, et al. Conditionally immortalized astrocytic cell line (BAS8.1) engineered to express glutamate decarboxylase (GAD65) synthesize and release GABA. Society for Neuroscience Abstracts. 1997; 23: 118.
- Thompson K. Genetically engineered neural cells with controllable GABA or DOPA production: a possible method to control side effects after transplantation. 2nd VA/UCLA Research Conference on Parkinson's Disease and Movement Disorders. 2004; 29.
- Thompson K, Anantharam V, Behrstock S, Bongarzone E, Campagnoni A, et al. Conditionally immortalized cell lines, engineered to produce and release GABA, modulate the development of behavioral seizures. Experimental Neurology. 2000; 161: 481-489.
- Thompson KW. Genetically engineered cells with regulatable GABA production can affect afterdischarges and behavioral seizures after transplantation into the dentate gyrus. Neuroscience. 2005; 133: 1029-1037.
- Thompson KW, Suchomelova LM. Transplants of cells engineered to produce GABA suppress spontaneous seizures. Epilepsia. 2004; 45: 4-12.
- Gernert M, Thompson KW, Loscher W, Tobin AJ. Genetically engineered GABA-producing cells demonstrate anticonvulsant effects and long-term transgene expression when transplanted into the central piriform cortex of rats. Exp Neurol. 2002; 176: 183-192.
- Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000; 24: 372-376.
- Shimozaki K, Nakashima K, Niwa H, Taga T. Involvement of Oct3/4 in the enhancement of neuronal differentiation of ES cells in neurogenesis-inducing cultures. Development. 2003; 130: 2505-2512.
- Cenci MA, Lee CS, Bjorklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci.1998; 10: 2694-2706.
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, et al. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur J Neurosci. 2002; 15: 120-132.
- Lane EL, Winkler C, Brundin P, Cenci MA. The impact of graft size on the development of dyskinesia following intrastriatal grafting of embryonic dopamine neurons in the rat. Neurobiol Dis. 2006; 22: 334-345.
- Torres EM, Dunnett SB. Amphetamine induced rotation in the assessment of lesions and grafts in the unilateral rat model of Parkinson's disease. Eur Neuropsychopharmacol. 2007; 17: 206-214.
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Sixth ed. San Diego, California: Elsevier Inc; 2007.
- Kemp K, Gordon D, Wraith DC, Mallam E, Hartfield E, et al. Fusion between human mesenchymal stem cells and rodent cerebellar Purkinje cells. Neuropathol Appl Neurobiol. 2011; 37: 166-178.
- Kemp K, Gray E, Wilkins A, Scolding N. Purkinje cell fusion and binucleate heterokaryon formation in multiple sclerosis cerebellum. Brain. 2012; 135: 2962-2972.
- Chu K, Kim M, Jung KH, Jeon D, Lee ST, et al. Human neural stem cell transplantation reduces spontaneous recurrent seizures following pilocarpine-induced status epilepticus in adult rats. Brain Res. 2004; 1023: 213-221.
- Black S, Kadyrov M, Kaufmann P, Ugele B, Emans N, et al. Syncytial fusion of human trophoblast depends on caspase 8. Cell Death Differ. 2004; 11: 90-98.
- Potgens AJ, Schmitz U, Bose P, Versmold A, Kaufmann P, et al. Mechanisms of syncytial fusion: a review. Placenta. 002; 23 Suppl A: S107-S113.
- Cselenyak A, Pankotai E, Horvath EM, Kiss L, Lacza Z. Mesenchymal stem cells rescue cardiomyoblasts from cell death in an in vitro ischemia model via direct cell-to-cell connections. BMC Cell Biol. 2010; 11: 29.
- Jin HK, Schuchman EH. Ex vivo gene therapy using bone marrow-derived cells: combined effects of intracerebral and intravenous transplantation in a mouse model of Niemann-Pick disease. Mol Ther. 2003; 8: 876-885.