
Review Article
J Stem Cell Res Transplant. 2016; 3(1): 1023.
Targeting Strategies of Cancer Stem Cells in the Management of Solid Tumors
Acheampong A and Mousa SA*
The Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, Rensselaer, NY, USA
*Corresponding author: Shaker A Mousa, The Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, NY, USA
Received: June 01, 2016; Accepted: July 25, 2016; Published: July 27, 2016
Abstract
Conventional anti-cancer therapy often fails to provide a complete cure. There is hope that study of Cancer Stem Cells (CSCs) will contribute to change this. CSCs have been identified in hematological malignancies, and as a concept CSCs continue to gain acceptance as a small subpopulation of tumor cells in tumors that are capable of self-renewal and differentiation. They maintain distinct properties that facilitate tumor initiation, growth, and the ability to metastasize. Tumor drug resistance, disease aggressiveness, and recurrence have been linked to the tumor microenvironment and the involvement of CSCs’ properties such as its surface proteins (e.g., CD133, CD44, and CD49f), aldehyde dehydrogenase-1 activity, and aberrant developmental signaling pathways (e.g., Wnt, Notch, Hedgehog, and Hippo). This review focuses on the role of these signaling pathways in CSC biology, its implications for solid tumors, and the significance of potentially targeting Hippo for effective CSC elimination or suppression, including using novel nanotechnology-based drug delivery systems as a functional platform for therapeutic improvement.
Keywords: Aldehyde dehydrogenase 1; Cancer stem cells; Hippo signaling; Nanomedicine; Solid tumors
Introduction
A theoretical model being used to account for the numerous challenges facing conventional cancer therapies, including labeling solid tumor malignancies as incurable, is the presence of specific tumor cell subpopulations in Cancer Stem Cell (CSC) theory. Action of these subpopulations of cells potentially drives tumor perpetuation, recurrence, and resistance to the damaging effects of traditional anticancer therapies. With the initial identification and success of CSCs in the treatment of leukemia, a parallel proposal that acknowledges the emergence of CSCs in solid tumors may play a rewarding part in the development and management of novel, targeted therapies designed to eradicate this subpopulation of cells [1]. Specific targeting of CSCs that selectively inhibit vital segments of various intrinsic signaling or specific cell surface markers is under preclinical and clinical investigation to provide researchers and clinicians with additional targets to increase therapy success and patient survival. Current investigative focus is on the role of CSCs in solid tumor progression, resistance to chemotherapy, disease relapse as well as the potential of targeting CSCs in the management of solid tumor.
Solid vs. Hematologic Malignancies
Solid tumors exist as abnormal tissue masses that grow in areas of the body other than the blood, bone marrow, or lymphatic cells and are usually devoid of any form of liquid or cysts. Such cancers in advanced stages of the disease may still gain the ability to spread to other organs through the process of metastatic tumor growth. Solid tumors are usually classified based on the cell type from which they originate [2]. About 80% of all cancers are commonly made up of tumors arising in tissues that include breast, colon, lung, ovary, and prostate [3], with the four basic tumor sites such as the breast, colorectal, lung, and prostate accounting for about 60% of all cancer cases in the United States [4]. Other solid tumors that occur less frequently but still show relatively high mortality rates are glioblastoma multiform and pancreatic ductal adenocarcinoma [3].
Hematological malignancies have been described as rare malignant disorders with deaths from lymphoid, hematopoietic, and related tissues. They account for about 6.2% of all deaths and have benefitted from new treatments obtained from prolific research and development centered mainly on such malignancies relative to solid tumors. Generally, it is much easier to obtain peripheral blood specimens of malignant cells from patients with hematological malignancies than to obtain specimens from solid tumors occurring in different body locations [5].
Solid Tumors and Heterogeneity
Solid tumors often develop properties that make them resistant to the current cytotoxic therapies available including chemotherapy and thereby render the majority of metastatic solid tumors incurable [6]. Based on physiology, solid tumors are made up of cancer cells and stromal cells and differ from normal tissues in a significant number of ways, mostly due to differences in vasculature. Unlike the regular, ordered vasculature of normal tissues, blood vessels occurring in tumors often present highly abnormal, distended capillaries with leaky walls and sluggish flow, and tumor growth requires the continuous growth of new vessels, called angiogenesis [7]. Cells within the solid tumor subpopulation have been found to display unmistakable functional diversity stemming from events of hyper-proliferation and increased genetic volatility that results in distinctive regions within the tumor with varied extents of proliferation, differentiation, vascularity, inflammation [8], and invasive capabilities. Tumors possessing heterogeneous tissues with abundant, phenotypically, and functionally distinct cell subpopulations also have different capacities to grow, metastasize, and develop drug resistance. Heterogeneity is therefore a defining feature of solid tumors [9,10].
Concept of Cancer Stem Cells
A model established as a cellular mechanism that contributes to heterogeneity in the majority of the different solid tumor cancer types is the Cancer Stem Cell (CSC) model. This was a direct result of the initial identification of leukemia-initiating stem cells by Bonnet and Dick for acute myeloid leukemia. They discovered that the cells were capable of establishing the disease in Non-Obese Diabetic/Severe Combined Immunodeficiency or Severe Combined Immunodeficiency (NOD/SCID or SCID) mice [9]. This model postulates that there is a hierarchical organization of cells from which only a small portion or subset becomes responsible for indefinitely driving and sustaining tumorigenesis and establishing the cellular heterogeneity inherent in the primary tumor [10]. This subset of cells, the CSCs, has been found to share many characteristics with normal stem cells including the ability to self-renew and give rise to differentiated progeny. There is growing evidence to suggest that CSCs can play a major role in cancer initiation, progression, resistance, recurrence, and metastasis of some selected cancers [11]. Tumors that do not follow a CSC model may also contain tumorinitiating cells, but these cells do not exhibit stem cell-like properties.
The quiescent nature of CSCs is directly associated with and relevant to cancer therapy. Accumulating evidence indicates that diverse solid tumors that are resistant to chemotherapy possess quiescent CSCs. Through the activity of intrinsic and extrinsic protective mechanisms that help with their maintenance and longevity, CSCs often confer tumor resistance to conventional chemotherapy and contribute to disease relapse when treatment is discontinued. This indicates the need to understand such mechanisms and to develop novel approaches that target this dormancy to therapeutically manipulate and ultimately develop strategies to permanently eliminate this population of cells. By specifically targeting these mechanisms, the dormant CSCs may potentially become activated and hence rendered susceptible to therapy [12].
Clinical Significance of Cancer Stem Cells
The concept of CSCs has drawn much attention because it provides an explanation for the development of resistance in solid tumors to current non-surgical cancer therapies, primarily chemotherapy and radiotherapy, by displaying similar phenotypes as multidrug-resistant cells that favor the expression of drug efflux transporters [13] and activation of anti-apoptotic signaling pathways [14]. This eventually leads to tumor relapse [10,11]. The concept continues to influence current approaches to cancer research and therapy [15]. It differs from the classical clonal evolution model because that theory typically lacks any form of association with a hierarchical organization [9]. Although current cancer therapies can kill the bulk of cancer cells and enhance the length of survival after diagnosis of cancer, often these therapies are unable to wipe out the CSCs. The CSCs survive and give rise to new tumors and metastases, resulting in disease relapse. Such recurring tumors increase in malignancy, metastasize at a much faster rate, and become resistant to previously used therapeutic drugs [15].
Yet, solid tumors have been found to exhibit plasticity and a dynamic phenotype such that targeting and eradication of solely CSCs without also eliminating other non-CSCs may still not result in a complete cure. This is mainly due to the increased ability of CSC tumor cells to reverse their phenotype from non-CSCs to become CSCs under certain conditions. For cancer therapy to be successful, both bulk non-CSCs as well as CSCs must be eradicated [16].
Biological Properties of Cancer Stem Cells
The CSC model continues to gain support and understanding due to many revolutionary research reports. An example is the use of aberrant embryonic signaling pathways by CSCs, a main focus of this review as a critical piece for maintaining tumor self-renewal. So far, research on CSCs has been done primarily using cancer cell lines, xenograft, and tissue samples from patients. Since the initial isolation of CSCs in solid tumors after their identification in breast cancer in 2003 by Al-Hajj and colleagues [17]. CSCs have also been isolated in a variety of other solid tumors including glioblastoma, gastric, lung, melanoma, prostate, and ovarian [18-23]. Each type of tumor produced CSCs that shared typical features such as the ability to propagate tumors and resistance to chemotherapies.
Tumorigenic subpopulation of CSCs has also been distinguished from the bulk non-CSC tumor population using a variety of common markers reported in the literature for solid tumors. It has been shown that specific cell populations of CSCs exist in different cancers and may be identified and characterized by specific cell surface markers that depend on the type of cancer. Although no universal CSC markers are known, various cell surface markers present either singly or in combination with other markers have proven to be useful tools for distinguishing between CSC and non-CSC populations in both established cell lines and human samples [24]. Common cell surface markers for CSC isolation include specific phenotypes or the expression pattern of cell-surface proteins that include but are not limited to CD44, CD49f, and CD133, in addition to cellular activities such as aldehyde dehydrogenase activity.
CSCs in Breast Cancer
Among the different surface cell markers that demonstrate the existence of CSCs in solid tumors, CD44 and CD133 have received the most consideration with regard to both mesenchymal and epithelial tumors and have been implicated in therapeutic drug treatment resistance. Transmembrane glycoprotein CD44 functions as an extracellular matrix receptor for cell adhesion and binds extracellular elements including the glycosaminoglycan hyaluronic acid. This marker assists in the attachment of CSCs to the matrix and has been linked with the proliferation and metastasis of malignant tumor cells [17]. Based on the findings of the pioneering study using a NOD/SCID mice model, breast cancer was established as the first solid malignancy to express CSCs when Al-Hajj et al. identified CD44+CD24-/low as a cell subpopulation that maintained a significantly elevated tumor-initiating capability [17]. Only a few number of cells with this phenotype provoked tumor formation that recapitulated the phenotypic heterogeneity of the primary tumor when the cells were implanted into NOD/SCID mice. Conversely it was confirmed that other phenotypically diverse cells carried different surface markers, although none of those cells retained the ability to form tumors [17].
Results from additional studies strengthened the role of CD44+CD24-/ low population as critical proponents in breast cancer metastasis [25].
CSCs in Brain Cancer
Glioblastoma multiform stem cells have demonstrated tumorinitiating abilities and have also been found to model highly invasive tumors that are extremely resistant to radiation and hence evade DNA damage following orthotopic implantation [26]. CD133 is a pentaspan transmembrane glycoprotein that was initially isolated from hematopoietic stem cells. It is restricted to membrane protrusions and microvilli and has been used in the identification of CSCs in brain, colon, liver, and other solid malignancies and sarcomas [27,28]. After identification of the CD44 marker in breast cancer cells, CD133 was used to identify cells with tumor-initiating capabilities in various solid malignancies. Results from Singh et al. [29] showed that by using CD133 as a cell surface marker CSCs could be characterized and isolated to prove their existence in human brain malignancies such as glioblastoma multiforme using NOD/SCID mice models. They reported that while as few as 100 CD133+ cells could produce a tumor that phenocopied the patient’s original tumor, 105 CD133- cells engrafted but did not produce the same response.
An example of promising therapeutic potential in conjunction with current therapeutics for glioblastoma are results that showed that targeting pluripotency transcription factors SOX2, OCT4, and/ or Nanog homeobox and their combination may be a way to achieve optimal management of glioma [30].
CSCs in Colon Cancer
A study in human colorectal adenocarcinoma cell lines HT29 and Caco2 to determine a novel but functional CSC cell surface marker for colorectal cancer showed that CD49f, also known as integrin a6 (ITAGA6) and that functions as a laminin receptor for cell adhesion, is important for identification of CSCs in colorectal cancer. CD49f+ cells localized in cell fractions of CD44+ and CD133+ such that CD133+ and CD44+ cells that were negative for CD49f exhibited no tumorigenic activity, and highly tumorigenic cells could be enhanced in samples that had higher CD49f expression [31-35]. Ultimately, CD49f expression was found to be associated with tumor cell invasion and metastasis by means of integrin-mediated cell signaling [32-35].
Aldehyde Dehydrogenase 1 Activity
Aldehyde Dehydrogenases (ALDHs) are a group of cytoplasmic enzymes responsible for the oxidization of toxic aldehydes to carboxylic acids during metabolism of alcohol, and their activity is usually used as a marker for the identification of high-risk patients with either lung or breast cancer. This detoxification function is critical for stem cell durability and is the most probable explanation for the evident resistance that CSCs exhibit to chemotherapies that produce toxic aldehyde intermediates [36]. A study done on normal and malignant mammary epithelial cells using the ALDEFLOUR assay to assess the presence and proportion of cells that exhibited ALDH enzymatic activity showed that cells isolated from normal mammary epithelial cells expressing the stem/progenitor cell marker ALDH activity also had phenotypic and functional characteristics of mammary stem cells. This was also the case for ALDEFLUORpositive cells isolated from human breast tumors containing the CSC subpopulation unlike ALDEFLOUR-negative cells. Using a NOD/ SCID mice model, it was confirmed that for ALDH1-positive breast tumors, there was a greater chance for the CSCs’ subpopulation to acquire the same defining properties of normal stem cells that conferred tumor aggressiveness such as the ability to self-renew, an elevated potential to proliferate, and chemoresistance [37].
CSCs’ Microenvironment
Similar to normal stem cells, CSCs also depend on their microenvironment because it affects their activity. For CSCs, the microenvironment acts as a source of protection and allows for the preservation of their quiescent and undifferentiated states in tissues. This protective and supportive niche helps CSCs escape the effects of cytotoxic agents for chemotherapy and retain the ability to differentiate and proliferate. Within this niche the CSCs interact with other cell types including inflammatory cells, vascular endothelial cells, and fibroblasts, all of which are commonly present in nonepithelial stromal cells of solid tumors. Evidence has also shown that the niche is a structure with specific features that not only contain a diversity of cells but also cytokine and signaling pathways [38-41].
Another contributing factor to the heightened tumorigenicity and multidrug resistance potential of CSCs presented by the microenvironment is the depletion of oxygen supply to tumors. There have been some reports that point to CSCs exploiting ways to survive hypoxic conditions. This feature activates transcriptional factors called Hypoxia-Inducible Factors (HIF) by regulating the expression of certain target genes that affect the development and proliferation of cancer cells, regulate apoptosis, and promote blood vessel formation [42,43]. The HIFs, once activated, have the ability to up-regulate genes and molecules in a number of signaling pathways involved in the maintenance of the microenvironment including the PI3K/Akt/ mTOR signaling pathway. HIFs also stimulate the action of certain enzymes that aid in DNA repair, which promotes tumor growth and aggressiveness and results in poorer clinical outcome [44].
Signaling Pathways in CSCs
For CSCs the process of self-renewal and differentiation has been described as one that involves various signaling pathways. It becomes critical then to develop therapeutic strategies directed at inhibiting such CSC survival pathways with the intention of mainly targeting CSCs for elimination. In numerous cancer types, the aberrant embryonic signaling pathways identified and specifically associated with the regulation of CSCs include but are not limited to the Wnt/β-catenin, Notch, Hippo, and Hedgehog. When the Wnt signaling pathway is turned on, the transcription factor β-catenin, which is usually deactivated and bound in a phosphorylated state in the cytoplasm, becomes dephosphorylated and can move into the nucleus of the cell. Once in the nucleus, certain target genes necessary for maintaining homeostasis of tissues and that play a critical role in embryogenesis and cancer development become activated [45]. Notch is another signaling pathway where inappropriate activation has been linked to the metastatic potential of CSCs and therapeutic drug resistance. It is also involved in angiogenesis, stimulates proliferation, and regulates self-renewal by restricting differentiation and apoptosis and ultimately promotes the survival of CSCs. There has been some preclinical success and progression into clinical phase for evaluation with agents that specifically target the Notch pathway (Table 1), but not for the Wnt pathway, which has proven to be a challenging target [46]. The Hippo signaling pathway has been described as one that regulates organ development and tissue regeneration through corresponding events that tend to affect cell proliferation, differentiation, and apoptosis by means of a kinase cascade in response to mechano-sensory and cell polarity inputs [47].
![]()
Signaling Pathway
Type of Cancer
Drug
Trial Phase
Identifier
Sponsor
Notch
Breast
MK0752
I
NCT00106145
Merck
Pancreatic
MK0752
I, II
NCT01098344
Cancer Research UK
Renal cell
RO4929097
II
NCT01141569
University Health Network, Toronto
Leukemia
PF-03084014
I
NCT00878189
Pfizer
Hedgehog
Solid tumors
GDC-0449
I
NCT00968981
Genentech
Colorectal
GDC-0449
II
NCT00636610
Genentech
Hematologic
PF04449913
I
NCT00953758
Pfizer
Basal cell
BMS833923
I
NCT00670189
Bristol-Myers Squibb
Medulloblastoma
LDE225
I
NCT00880308
Novartis
Table 1: Targeted signaling pathways Notch and Hedgehog and corresponding therapeutic drugs in clinical trials targeting cancer stem cells (Hippo signaling pathways are still in the preclinical stage).
Role of Hippo as a Tumor Suppressor Pathway
Although this pathway was initially discovered in the fruit fly Drosophila through studies that aimed to discover regulators of tissue growth, it has been an immense contribution to present knowledge of mammalian Hippo signaling (Figure 1). At the core of this evolutionally conserved phosphorylation kinase cascade in mammals are four components, namely the STE 20-like protein kinase 1 (MST1 and MST2) [48-51], the Salvador 1 (SAV1 or WW45) [52,53], MOBKL1A and MOBKL1B (together referred to as Mob1), and the large tumor suppressor homologues (LATS1 and LATS2) [54-56]. MST1/2 kinases physiologically form a complex with the adapter protein SAV1 on activation by upstream regulators. The complex in conjunction with Mob1 further phosphorylates and activates the downstream kinases LATS1/2. This results in inhibition of the homologous transcription co-activators Yes-associated protein or YAP and TAZ. These transcription factors are translocated from the nucleus and remain sequestered in the cytoplasm [57] by interacting with 14-3-3 proteins causing its proteasomal degradation and repression of growth [58].
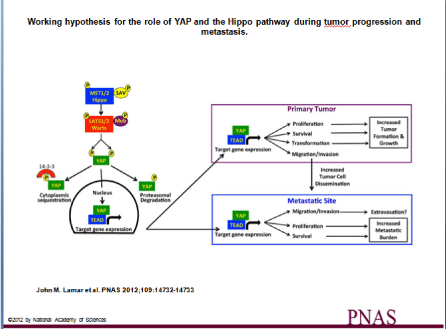
Figure 1: Working hypothesis for the role of YAP and the Hippo pathway during tumor progression and metastasis. The activation of the Hippo pathway by changes
in cell density, cell shape, and cell adhesion leads to phosphorylation of the Hippo kinases (MST1/2 in mammals), which in combination with the adaptor protein Sav
phosphorylate LATS1/2 kinases and their partner, MOB. The LATS/MOB complex then phosphorylates the transcriptional coactivator YAP and thereby represses
YAP activity by promoting both cytoplasmic sequestration via 14-3-3 proteins and proteasomal degradation. Our results show that inhibiting the ability of the Hippo
pathway to repress YAP results in increased YAP/TEAD-dependent gene expression, which influences both tumor growth and metastasis by enhancing processes
that occur at both the primary tumor and at the metastatic site. A close homolog of YAP, TAZ, is regulated in a similar fashion by the Hippo pathway and likely
plays a similar role.
Original Figure P1 from John M. Lamar et al. PNAS 2012; 109:14732-14733, copyright 2012 by National Academy of Sciences.
Under conditions where Hippo pathway is rendered inactive, the YAP/TAZ transcription factors do not become phosphorylated by LATS1/2, and this allows the accumulation of YAP/TAZ in the nucleus. There they interact with the DNA binding proteins TEAD belonging to the TEA domain family members. The interaction results in transcription of specific target genes associated with the regulation of differentiation, cell cycle, and apoptosis [59,60]. YAP/ TAZ can also bind other transcriptional factors including Smad1 [61], Smad2/3 [62], Smad7 [63], RUNX1/2 [64], p63/p73 [65], and ErbB4 [66,67] to produce similar results. The deregulation in part or whole of the pathway can cause tumorigenesis because it adversely affects the development of organs and the maintenance of homeostasis of adult organs [56].
With the knowledge gained from studies using knockdown mice models, the role of Hippo signaling in tumor suppression has been correlated with various human cancers stemming from the upregulation of either of the transcriptional factors YAP or TAZ. It has been found that tumor formation is the result of the uninterrupted over expression of YAP [68]. Up-regulation and nuclear localization of YAP levels were determined in a number of different cancers [69- 71], and it has also been revealed that mechano-transduction and YAP expression are a necessary requirement for the maintenance and generation of cancer-associated fibroblasts [72]. This information also provided details of the association between the over expression of the transcription factor TAZ and cell lines for non-small cell lung cancer and breast cancer cells [73,74].
Mutations and hypermethylation of upstream molecules of the core kinase proteins have been shown in some cases to produce malignant tumors. Genetically modified Merlin+/- mice have been found to develop malignant tumors such as hepatocellular carcinomas, lung adenocarcinomas, and fibrosarcomas. Merlin is a protein that functions upstream of Hippo cascade and induces the phosphorylation of LATS1/2 [75]. It is established that the consequences of deregulation or interruption of Hippo signaling activities clearly result in human cancers. Yet unlike in the case of the up-regulation of YAP/TAZ and its consequences, only a few germline and somatic mutations of Hippo signaling components have been identified [60,76,77]. This suggests that pharmacological inhibition of YAP and/or TAZ activity may be novel routes for anti-cancer therapeutic intervention.
A number of studies on gene expression profiling have shown the relevance of the transcription factors YAP/TAZ in CSCs by shedding more light on how elevated CSC subpopulations in breast cancer tissues overlap with gene expression induced by the YAP/ TAZ transcriptional factors [78]. YAP regulation has also been linked to this pathway through the regulatory action of miRNA-29 family because it inhibits the tumor suppressor PTEN, which then acts as an antagonist of PI3K, the upstream activator of mTOR. Eventually, by inactivating the PI3K/mTOR pathway, the activity of CSCs could be affected by Hippo signaling [79]. A study on the PI3K/mTOR pathway showed enhancement and reinforcement of breast CSCs obtained when the pathway was activated via knocking down PTEN [80].
For any therapeutic intervention to be successful, potentially targeting genes that are usually kept under control by the Hippo pathway should be a reasonable approach. So far the sole pre-clinical lead compound that targets Hippo signaling comes from studies addressing the YAP/TAZ interaction with the TEAD transcription factors [81]. However, other compounds have also been found to have potential for inhibiting transcriptional activity of YAP in vitro, such as verteporfin, which was found to be reasonably effective at blocking mouse YAP1-overexpression and is clinically used as a photosensitizer in photocoagulation therapy for macular degeneration [82]. Simvastatin is another drug that has been found to decrease nuclear activity of YAP, which interferes with protein geranylgeranylation and therefore arrests growth [83].
Role of Hedgehog Signaling
Activation of the Hedgehog signaling pathway leads to an increase in pro-angiogenesis factors, cyclins, and anti-apoptotic along with a decrease in apoptotic genes. Additionally, several reports demonstrated that the activation of Hedgehog pathway leads to increased fibrogenesis and carcinogenesis in many tissues including pancreatic tissue [84]. Furthermore, activation of Hedgehog signaling pathway regulates Epithelial-Mesenchymal Transition (EMT) in pancreatic cancer stem-like cells, which is implicated in pancreatic and non-small cell lung tumor progression, metastasis, and resistance [85,86]. Hedgehog signaling pathway has been also implicated in other types of cancer stem cell stemness and carcinogenesis including bladder, breast, and prostate cancer [87-89]. These data support the potential in targeting the Hedgehog signaling pathway in the management of various types of cancer (Table 1).
Impact of Nanotechnology in Targeting CSCs
A critical problem faced by most oncologists is the maintenance and preservation of non-cancerous host cells from the cytotoxic effects of conventional chemotherapeutic agents. A truly curative therapy would be one that targets not only the bulk of cancer cells but also CSCs. Thus there has been an increase in the drive to identify compounds that will provide a more effective action on CSCs but cause fewer side effects. There is also the need to develop new strategies for the administration of such novel compounds. With only a few drugs in our therapeutic arsenal that have demonstrated selective, high efficacy against CSCs such as salinomycin, it is important to focus on targeting CSCs in tumors. It would be beneficial to specifically target molecules in the Hippo pathway, which would also function precisely to eliminate CSCs. Nanomedicine and its drug delivery approaches are one avenue of great promise for targeting drug-resistant CSCs [42].
Nanotechnology makes the incorporation of multiple therapeutic, sensing, and target agents into delivery systems possible. Some of these include liposomes for the delivery of water-soluble drugs, dendrimers, gold nanoparticles, quantum dots, and micelles for water-insoluble drugs. The sizes of nano-materials used for loading target chemotherapeutic agents usually range between 1 and 100nm. Strategies for the construction of such anti-cancer nanoparticle complexes include encapsulation and covalent or noncovalent binding of components that facilitate the recognition or identification of the location of the cancer to deliver a therapeutic dose of an agent to kill the tumor cells [90-92]. Successful examples of approved nanomedicine for cancer therapy include liposomal doxorubicin Doxil®, albumin-bound paclitaxel Abraxane®, and PEG-L-Asparaginase Oncaspar®, all of which have been reported to have exceptional accumulation in tumors either by passive or active targeting [93].
Nanotechnology has so far shown significant promise in the areas of drug and drug delivery systems development. It has the potential to offer invaluable advances in not only oncology but also in many other branches of medicine such as cardiology, immunology, neurology, endocrinology, ophthalmology, pulmonary, orthopedics, and dentistry [93]. It has been useful in many nanomedicine capacities including diagnosing, treating, and preventing disease. It has also been helpful in regenerative medicine for the improvement of cell component interactions including the manipulation of cell proliferation and differentiation, and for cell maintenance and repair [94]. Given such a wide range of applications, nanotechnology can improve the bioavailability of an anti-cancer drug and decreases the levels of toxicity patients experience from chemotherapy [95], thus helping to overcome conventional therapy limitations by increasing the efficacy and safety of the delivery system [96].
Nanotechnology-based approaches have been applied to specifically target CSCs in diverse ways. Nano-carriers can be used to deliver anti-CSC agents that are insoluble and unstable, to label CSCs using their biological signatures such as their surface proteins, and finally as a standalone method to target and eliminate mainly CSCs without disrupting healthy stem cells [97-100].
Because many drug-resistant tumors and CSCs have exceptionally high amounts of surface proteins such as CD44, Wei et al. [99] developed nano-gel-drug conjugates as anti-cancer treatment specifically for drug-resistant tumors and CD44. The main focus for the design of the Cholesteryl-Hyaluronic Acid (CHA) nano-geldrug conjugates was enhancement of different facets of drug delivery such as the solubility of the drug, its release from the nano-gel once in the cytoplasm, and active tumor targeting. It was demonstrated that compared to simple HA-drug conjugates, CHA-drug showed higher affinity for cellular membrane than for CD44 receptors in drug-resistant cancer cells. This resulted in better cellular uptake and therefore stronger cytotoxicity of CHA-drug in both cancer cells and spheroids. They also showed that the CHA nano-gel-drug conjugates, although simple macromolecular drugs, could effectively be used as promising contenders for the treatment of CD44-expressing tumors and to target and eradicate CSCs in an effort to eliminate drugresistance to chemotherapy and subdue tumor relapse [99].
Conclusion and Future Prospects
Existence of CSCs in solid malignancies and their critical ability to undergo self-renewal and differentiation for tumor initiation, progression, metastasis, recurrence and ultimately maintaining tumorigenesis has been substantiated. To date various potential therapeutic agents against CSCs have been determined. These agents are aimed at the various mechanisms that help CSCs to survive. Mechanisms that have been targeted include surface biomarkers, detoxifying enzymes, and DNA repair enzymes. The CSC concept proposes that for any novel therapeutic agent to be deemed successful, it must be designed to target and eliminate the entire subpopulation of CSCs and not just the bulk of cancer cells. With the recent studies on the Hippo pathway and its exposure as a potential means of targeting CSCs, it is imperative to get a better understanding of the pathway and design new strategies for curative purposes. Nanotechnology may be one such platform.
The application of nanotechnology to drug delivery continues to have significant impact in medicine with more than 20 successful nanoparticle therapeutics in clinical use so far. This legitimizes nanotechnology as a platform that may improve and increase the therapeutic response of anti-cancer drugs in the effort to reduce cancer recurrence and treatment resistance. With continued research and development efforts, nanotechnology, which has already made its mark in the field of medicine, is expected to have specific impacts in therapeutic approaches that involve CSCs.
References
- Wang D-q, Zhu H-t, Liu Y-f, Yin R-g, Zhao L, Zhang Z-j, et al. Multi-Role of Cancer Stem Cell in Children Acute Lymphoblastic Leukemia. In: Clinical Epidemiology of Acute Lymphoblastic Leukemia - From the Molecules to the Clinic. In Tech. 2013; 75-85.
- Gavhane YN, Shete AS, Bhagat AK, Shinde VR, Bhong KK, Khairnar GA, et al. Solid Tumors: Facts, Challenges and Solutions. Int J Pharma Sci Res. 2011; 2: 1-12.
- Stem Cell Network. Cancers: Solid Tumor.
- Butcher L. Solid Tumors: Prevalence, Economics, and Implications for Payers and Purchasers. Biotechnol Healthc. 2008; 5: 20-21.
- Chizuka A, Suda M, Shibata T, Kusumi E, Hor A, Hamaki T, et al. Difference between Hematological Malignancy and Solid Tumor Research Articles Published in Four Major Medical Journals. Leukemia. 2006; 20: 1655-1657.
- Krishan A, Fitz CM, Andritsch I. Drug Retention, Efflux, and Resistance in Tumor Cells. Cytometry. 1997; 29: 279-285.
- Shah-Yukich AA, Nelson AC. Characterization of Solid Tumor Microvasculature: A Three-Dimensional Analysis Using the Polymer Casting Technique. Lab Invest. 1988; 58: 236-244.
- DeNardo DG, Andreu P, Coussens LM. Interactions between Lymphocytes and Myeloid Cells Regulate Pro- Versus Anti-Tumor Immunity. Cancer Metastasis Rev. 2010; 29: 309-316.
- Bonnet D, Dick JE. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates from a Primitive Hematopoietic Cell. Nat Med. 1997; 3: 730-737.
- Visvader JE, Lindeman GJ. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell. 2012; 10: 717-728.
- Clarke MF, Fuller M. Stem Cells and Cancer: Two Faces of Eve. Cell. 2006; 124: 1111-1115.
- Li L, Bhatia R. Stem Cell Quiescence. Clin Cancer Res. 2011; 17: 4936-4941.
- Singh A, Settleman J. Emt, Cancer Stem Cells and Drug Resistance: An Emerging Axis of Evil in the War on Cancer. Oncogene. 2010; 29: 4741-4751.
- Reya T, Clevers H. Wnt Signalling in Stem Cells and Cancer. Nature. 2005; 434: 843-850.
- Herrera VL, Colby AH, Tan GA, Moran AM, O'Brien MJ, Colson YL, et al. Evaluation of Expansile Nanoparticle Tumor Localization and Efficacy in a Cancer Stem Cell-Derived Model of Pancreatic Peritoneal Carcinomatosis. Nanomedicine (Lond). 2016; 11: 1001-1015.
- Kanwar JR, Samarasinghe RM, Kamalapuram SK, Kanwar RK. Multimodal Nanomedicine Strategies for Targeting Cancer Cells as Well as Cancer Stem Cell Signalling Mechanisms. Mini Rev Med Chem. 2016.
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc Natl Acad Sci USA. 2003; 100: 3983-3988.
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003; 63: 5821-5828.
- Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, et al. A Tumorigenic Subpopulation with Stem Cell Properties in Melanomas. Cancer Res. 2005; 65: 9328-9337.
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005; 65: 10946-10951.
- Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and Progenitor-Like Cells Contribute to the Aggressive Behavior of Human Epithelial Ovarian Cancer. Cancer Res. 2005; 65: 3025-3029.
- Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, et al. Identification of Gastric Cancer Stem Cells Using the Cell Surface Marker CD44. Stem Cells. 2009; 27: 1006-1020.
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and Expansion of the Tumorigenic Lung Cancer Stem Cell Population. Cell Death Differ. 2008; 15: 504-514.
- Magee JA, Piskounova E, Morrison SJ. Cancer Stem Cells: Impact, Heterogeneity, and Uncertainty. Cancer Cell. 2012; 21: 283-296.
- Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, et al. Most Early Disseminated Cancer Cells Detected in Bone Marrow of Breast Cancer Patients Have a Putative Breast Cancer Stem Cell Phenotype. Clin Cancer Res. 2006; 12: 5615-5621.
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature. 2006; 444: 756-760.
- Albers AE, Chen C, Koberle B, Qian X, Klussmann JP, Wollenberg B, et al. Stem Cells in Squamous Head and Neck Cancer. Crit Rev Oncol/Hematol. 2012; 81: 224-240.
- Terry J, Nielsen T. Expression of CD133 in Synovial Sarcoma. Appl Immunohistochem Mol Morphol. 2010; 18: 159-165.
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of Human Brain Tumour Initiating Cells. Nature. 2004; 432: 396-401.
- Seymour T, Nowak A, Kakulas F. Targeting Aggressive Cancer Stem Cells in Glioblastoma. Front Oncol. 2015; 5: 159.
- Haraguchi N, Ishii H, Mimori K, Ohta K, Uemura M, Nishimura J, et al. CD49f-Positive Cell Population Efficiently Enriches Colon Cancer-Initiating Cells. Int J Oncol. 2013; 43: 425-430.
- Kitayama J, Nagawa H, Tsuno N, Osada T, Hatano K, Sunami E, et al. Laminin Mediates Tethering and Spreading of Colon Cancer Cells in Physiological Shear Flow. Br J Cancer. 1999; 80: 1927-1934.
- Enns A, Gassmann P, Schluter K, Korb T, Spiegel HU, Senninger N, et al. Integrins Can Directly Mediate Metastatic Tumor Cell Adhesion within the Liver Sinusoids. J Gastrointest Surg. 2004; 8: 1049-1059; discussion 1060.
- Dydensborg AB, Teller IC, Groulx JF, Basora N, Pare F, Herring E, et al. Integrin alpha6Bbeta4 Inhibits Colon Cancer Cell Proliferation and c-Myc Activity. BMC Cancer. 2009; 9: 223.
- Robertson JH, Yang SY, Winslet MC, Seifalian AM. Functional Blocking of Specific Integrins Inhibit Colonic Cancer Migration. Clin Exp Metastasis. 2009; 26: 769-780.
- Moreb JS. Aldehyde Dehydrogenase as a Marker for Stem Cells. Curr Stem Cell Res Ther. 2008; 3: 237-246.
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell. 2007; 1: 555-567.
- Scadden DT. The Stem-Cell Niche as an Entity of Action. Nature. 2006; 441: 1075-1079.
- Sneddon JB, Zhen HH, Montgomery K, van de Rijn M, Tward AD, West R, et al. Bone Morphogenetic Protein Antagonist Gremlin 1 Is Widely Expressed by Cancer-Associated Stromal Cells and Can Promote Tumor Cell Proliferation. Proc Natl Acad Sci U S A. 2006; 103: 14842-14847.
- Fuchs E, Tumbar T, Guasch G. Socializing with the Neighbors: Stem Cells and Their Niche. Cell. 2004; 116: 769-778.
- Kenny PA, Lee GY, Bissell MJ. Targeting the Tumor Microenvironment. Front Biosci. 2007; 12: 3468-3474.
- Vinogradov S, Wei X. Cancer Stem Cells and Drug Resistance: The Potential of Nanomedicine. Nanomedicine (Lond). 2012; 7: 597-615.
- Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, et al. Aldehyde Dehydrogenase 1-Positive Cancer Stem Cells Mediate Metastasis and Poor Clinical Outcome in Inflammatory Breast Cancer. Clin Cancer Res. 2010; 16: 45-55.
- Wirthner R, Wrann S, Balamurugan K, Wenger RH, Stiehl DP. Impaired DNA Double-Strand Break Repair Contributes to Chemoresistance in HIF-1 Alpha-Deficient Mouse Embryonic Fibroblasts. Carcinogenesis. 2008; 29: 2306-2316.
- Rattis FM, Voermans C, Reya T. Wnt Signaling in the Stem Cell Niche. Curr Opin Hematol. 2004; 11: 88-94.
- Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D, et al. Preclinical Profile of a Potent Gamma-Secretase Inhibitor Targeting Notch Signaling with in Vivo Efficacy and Pharmacodynamic Properties. Cancer Res. 2009; 69: 7672-7680.
- Hiemer SE, Varelas X. Stem Cell Regulation by the Hippo Pathway. Biochim Biophys Acta. 2013; 1830: 2323-2334.
- Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst Ortholog, Hippo, Restricts Growth and Cell Proliferation and Promotes Apoptosis. Cell. 2003; 114: 457-467.
- Pantalacci S, Tapon N, Leopold P. The Salvador Partner Hippo Promotes Apoptosis and Cell-Cycle Exit in Drosophila. Nat Cell Biol. 2003; 5: 921-927.
- Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo Promotes Proliferation Arrest and Apoptosis in the Salvador/Warts Pathway. Nat Cell Biol. 2003; 5: 914-920.
- Wu S, Huang J, Dong J, Pan D. Hippo Encodes a Ste-20 Family Protein Kinase That Restricts Cell Proliferation and Promotes Apoptosis in Conjunction with Salvador and Warts. Cell. 2003; 114: 445-456.
- Kango-Singh M, Nolo R, Tao C, Verstreken P, Hiesinger PR, Bellen HJ, et al. Shar-Pei Mediates Cell Proliferation Arrest During Imaginal Disc Growth in Drosophila. Development. 2002; 129: 5719-5730.
- Tapon N, Harvey KF, Bell DW, Wahrer DC, Schiripo TA, Haber D, et al. Salvador Promotes Both Cell Cycle Exit and Apoptosis in Drosophila and Is Mutated in Human Cancer Cell Lines. Cell. 2002; 110: 467-478.
- Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila Tumor Suppressor Gene Warts Encodes a Homolog of Human Myotonic Dystrophy Kinase and Is Required for the Control of Cell Shape and Proliferation. Genes Dev. 1995; 9: 534-546.
- Xu T, Wang W, Zhang S, Stewart RA, Yu W. Identifying Tumor Suppressors in Genetic Mosaics: The Drosophila Lats Gene Encodes a Putative Protein Kinase. Development. 1995; 121: 1053-1063.
- Pan D. The Hippo Signaling Pathway in Development and Cancer. Dev Cell. 2010; 19: 491-505.
- Ramos A, Camargo FD. The Hippo Signaling Pathway and Stem Cell Biology. Trends Cell Biol. 2012; 22: 339-346.
- Codelia VA, Irvine KD. Hippo Signaling Goes Long Range. Cell. 2012; 150: 669-670.
- Yu FX, Guan KL. The Hippo Pathway: Regulators and Regulations. Genes Dev. 2013; 27: 355-371.
- Hong W, Guan KL. The YAP and TAZ Transcription Co-Activators: Key Downstream Effectors of the Mammalian Hippo Pathway. Semin Cell Dev Biol. 2012; 23: 785-793.
- Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, et al. Nuclear CDKs Drive Smad Transcriptional Activation and Turnover in BMP and TGF-beta Pathways. Cell. 2009; 139: 757-769.
- Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, et al. TAZ Controls Smad Nucleocytoplasmic Shuttling and Regulates Human Embryonic Stem-Cell Self-Renewal. Nat Cell Biol. 2008; 10: 837-848.
- Ferrigno O, Lallemand F, Verrecchia F, L'Hoste S, Camonis J, Atfi A, et al. Yes-Associated Protein (YAP65) Interacts with Smad7 and Potentiates Its Inhibitory Activity against TGF-beta/Smad Signaling. Oncogene. 2002; 21: 4879-4884.
- Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW Domain-Containing Yes-Associated Protein (YAP) Is a Novel Transcriptional Co-Activator. EMBO J. 1999; 18: 2551-2562.
- Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, et al. Physical Interaction with Yes-Associated Protein Enhances P73 Transcriptional Activity. J Biol Chem. 2001; 276: 15164-15173.
- Komuro A, Nagai M, Navin NE, Sudol M. WW Domain-Containing Protein YAP Associates with ErbB-4 and Acts as a Co-Transcriptional Activator for the Carboxyl-Terminal Fragment of ErbB-4 That Translocates to the Nucleus. J Biol Chem. 2003; 278: 33334-33341.
- Omerovic J, Puggioni EM, Napoletano S, Visco V, Fraioli R, Frati L, et al. Ligand-Regulated Association of ErbB-4 to the Transcriptional Co-Activator YAP65 Controls Transcription at the Nuclear Level. Exp Cell Res. 2004; 294: 469-479.
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell. 2007; 130: 1120-1133.
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, et al. Identification and Validation of Oncogenes in Liver Cancer Using an Integrative Oncogenomic Approach. Cell. 2006; 125: 1253-1267.
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007; 21: 2747-2761.
- Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, et al. Expression of Yes-Associated Protein in Common Solid Tumors. Hum Pathol. 2008; 39: 1582-1589.
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-Dependent Matrix Remodelling Is Required for the Generation and Maintenance of Cancer-Associated Fibroblasts. Nat Cell Biol. 2013; 15: 637-646.
- Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, et al. A Role for TAZ in Migration, Invasion, and Tumorigenesis of Breast Cancer Cells. Cancer Res. 2008; 68: 2592-2598.
- Zhou Z, Hao Y, Liu N, Raptis L, Tsao MS, Yang X. TAZ Is a Novel Oncogene in Non-Small Cell Lung Cancer. Oncogene. 2011; 30: 2181-2186.
- McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, et al. Mice Heterozygous for a Mutation at the Nf2 Tumor Suppressor Locus Develop a Range of Highly Metastatic Tumors. Genes Dev. 1998; 12: 1121-1133.
- Harvey KF, Zhang X, Thomas DM. The Hippo Pathway and Human Cancer. Nat Rev Cancer. 2013; 13: 246-257.
- Johnson R, Halder G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat Rev Drug Discov. 2014; 13: 63-79.
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell. 2011; 147: 759-772.
- Strassburger K, Tiebe M, Pinna F, Breuhahn K, Teleman AA. Insulin/IGF Signaling Drives Cell Proliferation in Part Via Yorkie/YAP. Dev Biol. 2012; 367: 187-196.
- Kolev VN, Wright QG, Vidal CM, Ring JE, Shapiro IM, Ricono J, et al. PI3K/mTOR Dual Inhibitor VS-5584 Preferentially Targets Cancer Stem Cells. Cancer Res. 2015; 75: 446-455.
- Gomez M, Gomez V, Hergovich A. The Hippo Pathway in Disease and Therapy: Cancer and Beyond. Clin Transl Med. 2014; 3: 22.
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and Pharmacological Disruption of the TEAD-YAP Complex Suppresses the Oncogenic Activity of YAP. Genes Dev. 2012; 26: 1300-1305.
- Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, et al. Interplay of Mevalonate and Hippo Pathways Regulates RHAMM Transcription Via YAP to Modulate Breast Cancer Cell Motility. Proc Natl Acad Sci U S A. 2014; 111: E89-E98.
- Bai Y, Bai Y, Dong J, Li Q, Jin Y, Chen B, et al. Hedgehog Signaling in Pancreatic Fibrosis and Cancer. Medicine (Baltimore). 2016; 95: e2996.
- Wang F, Ma L, Zhang Z, Liu X, Gao H, Zhuang Y, et al. Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem-Like Cells. J Cancer. 2016; 7: 408-417.
- Bai XY, Zhang XC, Yang SQ, An SJ, Chen ZH, Su J, et al. Blockade of Hedgehog Signaling Synergistically Increases Sensitivity to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer Cell Lines. PLoS One. 2016; 11: e0149370.
- Syed IS, Pedram A, Farhat WA. Role of Sonic Hedgehog (Shh) Signaling in Bladder Cancer Stemness and Tumorigenesis. Curr Urol Rep. 2016; 17: 11.
- Fu YZ, Yan YY, He M, Xiao QH, Yao WF, Zhao L, et al. Salinomycin Induces Selective Cytotoxicity to MCF-7 Mammosphere Cells through Targeting the Hedgehog Signaling Pathway. Oncol Rep. 2016; 35: 912-922.
- Suzman DL, Antonarakis ES. Clinical Implications of Hedgehog Pathway Signaling in Prostate Cancer. Cancers (Basel). 2015; 7: 1983-1993.
- Sajja HK, East MP, Mao H, Wang YA, Nie S, Yang L. Development of Multifunctional Nanoparticles for Targeted Drug Delivery and Noninvasive Imaging of Therapeutic Effect. Curr Drug Disc Technol. 2009; 6: 43-51.
- Srinivasan M, Rajabi M, Mousa SA. Multifunctional Nanomaterials and Their Applications in Drug Delivery and Cancer Therapy. Nanomaterials. 2015; 5: 1690-1703.
- Rajabi M, Srivnivasan M, Mousa SA. Nanobiomaterials in Drug Delivery. In: Grumezescu A (eds). Nanobiomaterials in Drug Delivery. Applications of Nanobiomaterials. 9. Elsevier, Amsterdam. 2016; 1-38.
- Farokhzad OC, Langer R. Nanomedicine: Developing Smarter Therapeutic and Diagnostic Modalities. Adv Drug Del Rev. 2006; 58: 1456-1459.
- Patil M, Mehta DS, Guvva S. Future Impact of Nanotechnology on Medicine and Dentistry. J Indian Soc Periodontol. 2008; 12: 34-40.
- Petros RA, DeSimone JM. Strategies in the Design of Nanoparticles for Therapeutic Applications. Nat Rev Drug Discov. 2010; 9: 615-627.
- Sahay G, Alakhova DY, Kabanov AV. Endocytosis of Nanomedicines. J Control Release. 2010; 145: 182-195.
- Lee K, Drachev VP, Irudayaraj J. DNA-Gold Nanoparticle Reversible Networks Grown on Cell Surface Marker Sites: Application in Diagnostics. ACS Nano. 2011; 5: 2109-2117.
- Sadhukha T, Niu L, Wiedmann TS, Panyam J. Effective Elimination of Cancer Stem Cells by Magnetic Hyperthermia. Mol Pharm. 2013; 10: 1432-1441.
- Wei X, Senanayake TH, Warren G, Vinogradov SV. Hyaluronic Acid-Based Nanogel-Drug Conjugates with Enhanced Anticancer Activity Designed for the Targeting of CD44-Positive and Drug-Resistant Tumors. Bioconj Chem. 2013; 24: 658-668.
- Liu Y, Lu WL, Guo J, Du J, Li T, Wu JW, et al. A Potential Target Associated with Both Cancer and Cancer Stem Cells: A Combination Therapy for Eradication of Breast Cancer Using Vinorelbine Stealthy Liposomes Plus Parthenolide Stealthy Liposomes. J Control Release. 2008; 129: 18-25.