
Review Article
J Stem Cell Res Transplant. 2017; 4(1): 1025.
Deciphering the Cancer Puzzle: Cancer Stem Cells Being the Pivotal Piece
Basak U#, Dutta A#, Dutta Chowdhury D, Bhattacharya A, Banerjee S, Khan P and Das T*
Division of Molecular Medicine, Bose Institute, P-1/12 CIT Scheme VII M, Kolkata, India #These authors contributed equally to this work
*Corresponding author: Tanya Das, Division of Molecular Medicine, Bose Institute, P-1/12 CIT Scheme VII M, Kolkata 700 054, India/p>
Received: October 13, 2016; Accepted: January 06, 2017; Published: January 09, 2017
Abstract
Cancer is the most devastating concern of today’s world, being the highest cause of global mortality with millions of new detected cases. Despite thorough research and implementation of novel treatment strategies, the worldwide burden of cancer is steadily increasing. The tumor recurrence remains the most dreaded reason behind this vicious battle. Recently a small population of cells, adorned with the property of self-renewal, differentiation and capable of forming tumor in nude mice, was identified within the tumor mass. This subset of stemlike cells within the neoplastic tissue is designated as Cancer stem cells or tumor initiator cells. Extensive research has now indicated that the cancer stem cells are the major culprit behind tumor initiation, angiogenesis, invasion, metastasis, drug resistance and tumor relapse. In addition to differentiating into non stem tumor cells to drive tumor progression, evidences are being accumulated that they can even trans differentiate into non tumor cells of different lineages in order to support tumor maintenance and advancement. All these concepts back up the idea that cancer can be regarded as a ‘stem cell disease’ with cancer stem cells being the most potential therapeutic target. Moreover, the concept of tumor cell plasticity depicting the inter-conversion between the non-stem cancer cells into cancer stem cells and vice-versa opens a new era of cancer research and a huge possibility towards developing more successful treatment strategies.
Keywords: Cancer stem cells; Differentiation; Self-renewal; Dedifferentiation; Plasticity; Transdifferentiation
Abbreviations
ABC: ATP Binding Cassette; BMP: Bone Morphogenetic Protein; bCSCs: breast CSCs; CAFs: Cancer Associated Fibroblasts; CSCs: Cancer Stem Cells; CNS: Central Nervous System; CTLA4: Cytotoxic T-Lymphocyte-Associated Antigen4; ECs: Endothelial Cells; ELCs: Endothelial Like Cells; EGF: Epidermal Growth Factor; EMT: Epithelial to Mesenchymal Transition; GBM: Glioblastoma Multiforme; GSCs: Glioma Stem Cells; iPSCs: Induced Pluripotent Stem Cells; IL: Interleukin; MMP: Matrix Metalloproteinase; NSCCs: Non-Stem Cancer Cells; PD1: Programmed Cell Death Protein 1; PD-L1: PD-ligand-1; TF: Transcription Factor; TGF: Transforming Growth Factor; TAMs: Tumor-Associated Macrophages; TANs: Tumor Associated Neutrophils; VEGF: Vascular Endothelial Growth Factor
Introduction
Cancer figure amongst the most devastating causes of morbidity and mortality globally. According to the World Health Organization (WHO), approximately 8.2 million deaths occurred due to cancer in 2012 while 14 million new cases were detected during the same period [1] and the number of new cases is expected to increase by 70% over the next 2 decades. This depressing statistic may be due to high rate of cancer recurrence which does not permit the disease-free survival of the patients. Recent studies have designated cancer stem cells, which are responsible for the tumor initiation, maintenance and metastasis [2], as the sole contributors of the various factors and traits associated with tumor aggression, resistance and relapse [3-5]. According to the American Association of Cancer Research, a CSC has been defined as ‘a cell within a tumor that possesses the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor’ [6] (Figure 1). In fact, like normal Stem Cells (SCs), CSCs are able to sustain multi-lineage differentiation and self renewal properties. The CSC concept can find its origins way back in the 19th century when the German pathologist Rudolf Virchow postulated that cancers occur because of the aberrant activation of dormant embryonic cells in the adult tissue [7]. He further implied that cancer does not originate spontaneously but that cancer cells must arise from other living cells. However, it was only after another 100 years when Dick et al. [8] in 1994 isolated leukemia SCs that the first concrete evidence supporting the CSC hypothesis arose. They defined these stem-like cells as leukemia initiating cells capable of forming tumor in nude mice. Subsequently, brain CSCs were discovered in 2001 [9,10] following which CSCs have now been discovered in the tumors of various different tissues and organs like breast [11], lung [12,13], colon [14-16], pancreas [17,18] ovary [19] and in melanoma [20]. Studies are underway trying to identify and characterize CSCs in other tissues as well. Here, we review the role of CSCs in cancer development and therapy as the evidence in support of the hypothesis that cancer is a stem cell disease.
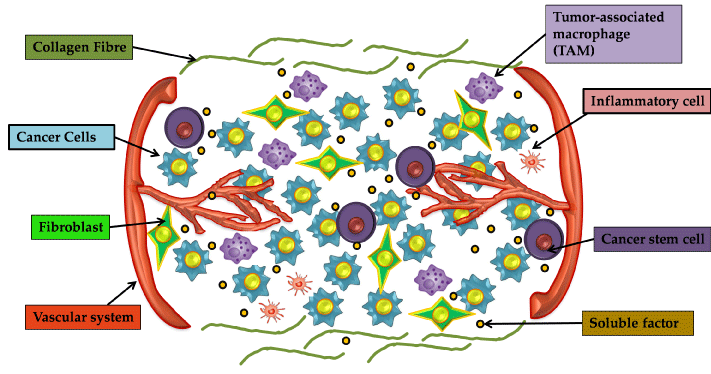
Figure 1: The various components of tumor microenvironment contributing to the intratumoral heterogeneity. The tumor micoroenvironment in addition to CSCs
and NSCCs, comprises of various inflammatory cells, tumor associated macrophages, fibroblasts etc which cooperatively orchestrate the tumor survival.
Contribution of Cancer Stem Cells in the Progression of Cancer
Tumor initiation
A normal tissue maintains its homeostasis by the function of the somatic stem cells that tightly regulates tissue parameters. In the case of a tumor, these parameters are dysregulated and disrupted which ultimately leads to the neoplastic transformation. The initial conditions faced by developing neoplastic cells are very hostile for its continued survival. Even though the immune cells try and destroy this growing abnormality within an individual with extreme prejudice, however, they are not always successful and on the rare occasion, these neoplastic cells are able to escape immune regulation. In addition, a subpopulation of cells in a tumor mass shows high drug resistant properties that allow them to survive and proliferate. These cells are termed as Cancer Stem Cells (CSCs). An increasing number of reports and evidence show that tumor-propagating cells stem from a small population of undifferentiated cancer cells with stem like properties [21] (Figure 2). In fact, considering the ability of CSCs to differentiate into heterogeneous non-stem cancer cell population that creates a tumor mass, it is logical to conceptualize the contribution of CSCs in tumor initiation. Reports indicate that stem cells are associated with the initiation and progression of cancer [22]. Breast CSCs with CD44 (hi)/CD24 (lo) phenotype have been represented as the main driving factor in breast cancer initiation [23]. Boumahdi et al. [24] discovered the invaluable role of SOX2 in regulating tumor initiation and CSC properties. Recent reports also demonstrate that existence of CSCs in the brain that are responsible for the intractability of GBM and its initiation [25]. MSCs have also been implicated in enhancing lung cancer initiation through secretion of IL-6 [26]. In fact, co-culture of lung cancer cells, CL1-5 and A549, with MSCs is noted to enhance the ability of tumor cells to over-express pluripotency markers, form tumorospheres, and show enhanced drug resistance. These results imply the role of MSCs in stimulating CSCs, thereby potentiating tumor initiation in immunodeficient mice by lung cancer cells [26].
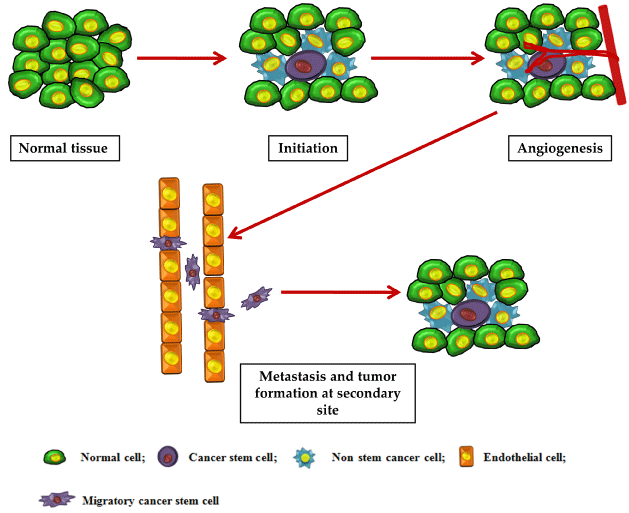
Figure 2: The progression of cancer generally occurs in a sequential manner. This figure depicts the role played by CSCs in tumor initiation, angiogenesis, invasion
and metastasis.
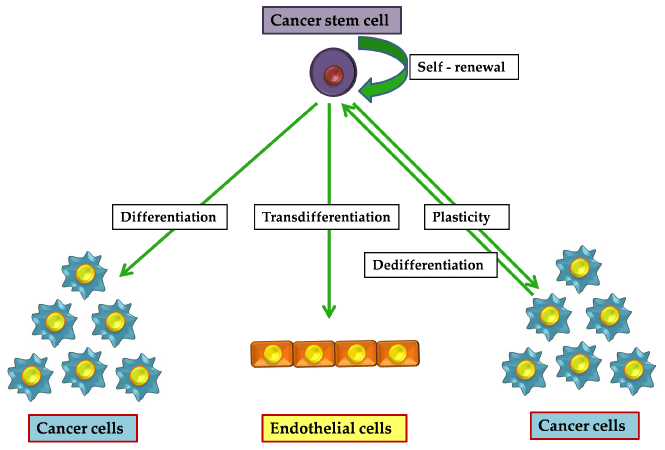
Figure 3: The paradigm of CSC function. The CSCs apart from maintaining their own pool by self-renewal, differentiate into NSCCs and can transdifferentiate into
cells of other lineages. Also, within the tumor the cells occur in a plastic state leading to interconversion between CSCs and NSCCs.
Tumor angiogenesis
Angiogenesis is a critical facet in the development and progression of tumors. They are able to stimulate juxtacrine cells to secrete proangiogenic factors to stimulate the formation of new blood vessels for nutrients and oxygen. In fact, once the tumor volume reaches 3mm3, the neoplastic growth becomes dependent on blood supply for which neo-angiogenesis or new blood vessel formation is of utmost necessity. Moreover, for its metastatic dissemination, the tumor mass requires angiogenesis. Tumor angiogenesis is triggered not only by up-regulation of angiogenic factors but also by down-regulation of anti-angiogenic molecules [27] (Figure 2). Recently the role of CSCs in tumor angiogenesis has caught a lot of attention. Hypoxia has a major role in CSC induced angiogenesis since experiments have shown that hypoxia enhances the CSC population in a tumor [28]. Recently, the role of hypoxia in regulating the self-renewal property of CSCs by enhancing the activity of stemness proteins like Oct- 4,Sox-2 and Nanog has been reported [29,30]. It is believed that hypoxia induces VEGF secretion from CSCs triggering angiogenesis though the exact mechanism is not clear [31]. It is also important to note that CSCs survive various antiangiogenic therapies like Avastin (monoclonal antibody targeting VEGF-A) and Sunitinib (VEGFR inhibitor) due to the overexpression of drug effluxers, causing tumor relapse [32]. Now it is widely believed that by targeting CSCs, tumorangiogenesis can be inhibited. In fact, one of the critical pathways of CSCs and angiogenesis is BMP signaling [33]. While BMP-9 suppresses VEGF expression by the BMP-9/ALK1 pathway, TGFβ1/ ALK5 pathway enhances VEGF expression and angiogenesis [34].
The balance between these two pathways is maintained by BMP4 [34]. Interestingly, according to Piccirillo et al. [35], the tumorigenic potential of glioblastoma CSCs is impeded by BMP4 through BMP- 4/BMPR/SMAD signaling pathway. This discussion indicates the vital role of BMPs in tumorigenesis and angiogenesis by CSCs. It has also been observed that Notch inhibitor DAPT, which is a γ-secretase inhibitor, reduces CD133+ population of GSCs as well as their self-renewal ability [36]. However, more reports from research experiments are required to develop more efficient ways to inhibit CSC-induced angiogenesis.
Tumor invasion
Invasion and metastasis are two major hurdles for successful cancer therapy. After initiation, tumor cells go through Epithelial to Mesenchymal Transition (EMT) to be able to invade the surrounding stroma and the basement membrane in order to spread throughout an individual via the circulatory or lymphatic systems. In fact, during EMT, immotile epithelial cells, embedded in a tumor cell mass, dissolve their cell-cell junctions and get transformed into individual, motile and invasive mesenchymal cells [37]. Recent reports have correlated EMT with stemness gain of cancer cells [38,39] thereby implying the involvement of CSCs in tumor invasion (Figure 2). In fact, recent reports implicate that breast CSCs are endowed with aggressive invasion and migration property due to its intrinsic suppression of E-cadherin, which suppresses tumor formation [40].
For invasion, extracellular matrix proteolysis is of primary requirement [41]. Matrix Metalloproteinase (MMP) enzymes play essential role in the proteolytic degradation of extracellular matrix, thus leading to enhanced tumor invasion [42]. MMPs are a multigene family of nine or more highly homologous zinc-dependent endopeptidases that remodel ECM, alter cell-cell and cell-ECM interactions thereby playing a crucial role in the multistep process of invasion and metastasis. Among the MMPs, MT1-MMP, MMP-2 and MMP-9 [42] have been shown to be involved in tumor invasion both in vitro and in vivo nude mice model. Degradation of the cell junction proteins Occludin and Claudin by MMP 2 and MMP 9 sponsors the invasion of CSCs into the CNS. CSCs have also been reported to contribute to glioblastoma invasiveness and repopulation [43]. Invasion was dramatically diminished by knocking-down Netrin-1, an axon guidance molecule, from glioblastoma SCs [44]. However, more studies are required to fully elucidate the role of CSCs in tumor invasion.
Metastasis
Metastasis is the major complexity associated with poor prognosis and high mortality observed in cancer. It is a multistep process that begins with local invasion, EMT, intravasation into the circulatory system, extravasation out of the circulatory system, colonization and rehabilitation at a distant site [45] (Figure 2). Colonization encompasses a series of events which includes the continued existence of the cancer cells until they enter secondary tissue, form micrometastases followed by a latency period and regrowth, recirculation and the formation of lesions [46]. Involvement of CSCs in metastasis is well-accepted. In fact, heightened expression of metastasis-related genes in CD44+CD24low/- breast CSCs has been found to cause lung metastasis in vivo [47]. Osteopontin and hyaluronan, which are the chief components of the target tissues of breast cancer, e.g., liver, lung, bone and brain, serve as specific ligands for CD44 [48]. Osteopontin is involved in immune responses and cell adhesion and is also associated with an increased incidence of tumor invasion and metastasis [49]. In another study, CD133+ovarian CSCs that are accountable for the tumor metastasis and patients’ survival have been shown to promote tumor invasion and metastasis via the up-regulation of MMP-9 [50]. On the other, overexpression of CXCR4, IL-1 and osteopontin increases the migration potential of breast CSCs [51]. Also it was observed that CSCs possess higher migration potential than NSCCs due to the inherent subduing effect on the tumor suppressor, E-cadherin [40]. As has already been stated, the microenvironment of CSCs with all its cellular and molecular factors, play a vital role in metastasis. In a recent report we have shown that intrinsic non-migratory CXCR4- CSCs secrete soluble bio-modulators like EGF, that act in paracrine manner on Non-Stem Cancer Cells (NSCCs) to convert them to migratory CXCR4+ CSCs, thus assisting in metastasis and tumor recurrence in breast cancer patients [52]. In fact, unlike the intrinsic non-migratory CXCR4- CSCs that reside in the inner tumor mass, the highly migratory metastatic CXCR4+ CSCs reside in the peripheral/disseminating regions of the breast tumor. These results validate the spatiotemporal regulation of different stages of CSCs [52].
The role of CSCs in intra-tumoral heterogeneity – CSC differentiation
The hierarchy model places CSCs at its apex and nontumorigenic cancer cells differentiate from them [53,54] (Figure 3). Reports demonstrated that differentiative capacity exists within SCID leukemic SCs in vivo, which differentiate into leukemic blast cells [55]. When transplanted in immune-compromised mice, CD34+CD38- tumorigenic cells led to the development of CD34- and CD38+ cells thus exhibiting the differentiation ability of leukemic SCs [55,56]. Similarly, CD44+/CD24-/low breast CSCs upon transplantation gave rise to diverse phenotypic cells, e.g., CD 44+/CD24+ which has reduced proliferative and non-tumorigenic capacity and the tumorigenic population [11,56]. CD44+/a2β1/CD133+ prostate CSCs differentiated to bring up all surface markers as present in the primary tumor [57]. Similarly, the secondary tumor recapitulated the phenotypic diversity of primary tumor in ovarian cancer [58] and pancreatic cancer [17].
During multi-lineage differentiation, colon CSCs are present at various stages of differentiation since it developed to gobletlike, enterocyte-like and neuroendocrine-like cells in vivo [59]. In Ewing’s sarcoma, transplantation of CD133+ subpopulation showed differentiation to adipogenic, osteogenic and chondrogenic lineages in vitro [60]. In fact, besides giving rise to multi-lineage tumor cells, CSCs also transdifferentiate into non-tumor cells of different lineages such as vascular endothelial cells and pericytes [61]. This discussion opens door for so many research opportunities and therapy targets.
The basic causes behind the heterogeneity in tumor are epigenetic changes [56] and the changes in the signaling pathways like Notch, Hedgehog and Wnt [61]. In case of the lineage-determining steps of colon CSCs, notch signaling decides whether goblet-like or enterocyte-like program will be followed [59,62]. PI3K inhibitors induce differentiation of CSCs, thereby, making PI3K responsible for stemness [59]. Multi-lineage differentiation of liver CSCs is due to repression of Oct4, Sox2, Nanog, Lin-28, c-myc and Klf-4. Mainly, Oct4 is responsible [63], which in turn, can be regulated by the tumor microenvironment [63,64]. BMP is highly expressed in glioma microenvironment with Gremlin 1, an antagonist of BMP, maintains the relationship between differentiation and proliferation of Glioblastoma CSCs [61,65]. Also, the differentiated glioblastoma cells secrete IL-6 which maintains CSCs [65]. Paneth cells provides favorable microenvironment for the sustenance of the Lgr+ intestinal SCs by releasing TGFa, EGF, WNT3 and Notch ligand Dll4 [66]. The tumor niche consisting of stromal myofibroblasts secretes factors which control Wnt cascade in colon CSCs [67]. The cancer niche also contains CAFs, MSCs, TAMS, TANs and extracellular matrix, affecting the course of CSC development [68] (Figure 1). miRNAs, which affect signaling pathways, can be a useful tool to alter the CSC characteristics [69].
To sum up, proliferation and differentiation of CSCs are regulated by numerous signaling pathways, factors influencing multi-lineage differentiation and its microenvironment, dictating about which fate to uptake.
Now that, we have addressed the ability of CSCs to differentiate, it is of paramount importance to question: how CSCs maintain their own pool if they differentiate into other cell types?
How Does CSCs Maintain their Pool?
Self renewal
It is evident that CSCs are able of giving rise to differentiate tumor cells and to replenish their own population so that they can perpetuate indefinitely [70] (Figure 3). This self-renewal property assures the continuous maintenance of CSC pool during tumor progression and initiation. The proof of self renewal in CSCs was first observed in leukemic SCs and was verified when xenotransplantation was carried out and subpopulation of tumor initiating cells were found even after serial transplantation [55]. The ability to recapitulate and form the tumor initiating sub-population was also found in breast [56], prostate [57] ovarian [58] and pancreatic [17] cancers. However, it is interesting to note that pathways followed for self renewal by normal SCs are deregulated in case of CSCs [71].
The hedgehog pathway plays a vital role in terms of self renewal as most of the genes involved in its regulation are oncogenes such as Smo, Shh, Gli1, Gli2 and Ptch1. Mutational activation of these genes, leading to activated hedgehog is the root cause of numerous cancers [72]. Mutation in tumor suppressor gene Ptch1can form breast cancer [71,72]. But mutation is not the only driving force of self renewal and CSC formation, overexpression of ligands also escalates the number of mammosphere-initiating cells. Furthermore, Gli1 and Gli2, which are transcriptional factors and positive mediators in this pathway, also experience a surge and potentiate hedgehog signaling [73]. As an aftermath, Bmi1, belonging to the polycomb gene family and a major regulator of self renewal in CSCs, is activated. Bmi1 is a transcriptional repressor which silences p16 INK4a and p19 Arf and is overexpressed in CSCs than non-SCs. Blocking Bmi1 abrogates hedgehog pathway and eventually self renewal of CSCs [73,74]. Self renewal is also tightly regulated by Notch and Wnt pathways and has an immense overall regulation by the niche of CSCs [74].
For the complete elimination of CSCs, self renewal pathways need to be targeted. However, the same pathways are followed in normal SCs. Hence, it is very vital to design drugs to specifically eliminate CSC sparing the normal SCs.
Dedifferentiation of NSCC to CSC
CSCs are capable of self renewal and causes tumor proliferation, invasion and are drug-resistant [75]. But, the differentiated cancer cells have limited proliferation capability and unable to initiate tumor, hence, it is more advantageous if CSC pool is maintained for tumorigenic growth. To achieve this, cancer cells undergo dedifferentiation process, under which they are reprogrammed genetically or epigenetically to gain the undifferentiated stem cell state [61] (Figure 3). Induction of EMT in mammary epithelial cells and mammary carcinomas express markers which are very similar to the markers presented by stem cell like cultured cells from mammary epithelium [76]. Breast cancer cells upon knockdown of E-cadherin, promotes EMT transition, which showed excellent mammosphere formation, drug resistance and tumor initiation capacity identical to CSCs [77,78]. Differentiated astrocytes, on oncogenic transduction, also lead to GSCs [79]. NF-κB, an important regulator in the inflammatory tumor microenvironment, activates Wnt signaling, resulting in the dedifferentiation of intestinal epithelial cells into tumorigenic cells [80]. Also, disabling of Akt and Ink4a with constitutive activation of EGF receptor directs astrocyte dedifferentiation towards glioma CSCs [81]. Dedifferentiation of NSCCs to CSCs during chemotherapeutic drug treatment [82] and metastasis [52] further support the notion that CSC hierarchical model is bidirectional in nature. However, it is unclear that if cancer cells can dedifferentiate into CSCs, then what stops it from doing the same when non-tumorigenic cells are transplanted in immunocompromised mice. Hence, it leads us to speculate about the conditions and mechanisms involved to drive the dedifferentiation process.
EMT transitions is one of the most important contributing factors for dedifferentiation [76-78] and it is regulated by ZEB1, Slug, Snail and Twist, which act as repressors for epithelial adhesion [83]. ZEB1 TF subdues the expression of miR-200b [84], resulting in activation of Bmi-1 [85], which promotes CSC formation and self renewal [83,85]. miRNA 200 family strongly suppresses the tumor formation ability in breast cancer in vivo, inactivating which encourages tumor progression [85]. MiR-200b is also known to inactivate the expression of Suz12, a histone modifying enzyme, accountable for the repression of E-cadherin along with ZEB1 and Snail. E-cadherin is involved in cell-cell adhesion and its suppression is necessary for inducing stemness [83]. So it can be concluded from above that miR-200b negatively regulates tumor formation and seeding capacity, and hence, inhibition of it is mandatory for dedifferentiation process. Wnt signaling, which is known to maintain CSCs, also positively regulates EMT. Wnt-β-catenin signaling also regulates the expression of Snail TF, involved in triggering of EMT by suppression of E-cadherin that negatively controls the Wnt pathway [83]. Consequently, they all form an inter-related pathway in which positive induction of Snail downregulates E-cadherin and up-regulates EMT, and as an outcome, it causes activation of Wnt pathway and hence further increasing EMT progression [83]. In addition, NF-κB signaling stabilizes β catenin and in turn, activates Wnt pathway, also contributes to the dedifferentiation strongly [75].
Genetic insults such as KRAS mutations leading to its activation along with transcription factor, Myc, thereby encouraging dedifferentiation and self renewal of pancreatic cancer cells [75,86]. Niche plays an indispensable part in implicating dedifferentiation. To cite an example, stromal myofibroblasts constitute a niche around colon cancer cells promoting its dedifferentiation by activating Wnt pathway [67]. TGF β growth factor along with sonic hedgehog, notch and EGF augments the EMT promotion by acting as external cues from the niche [75]. Additionally, external stimuli such as hypoxia inducible factors also provide ground for CSC development via EMT [87]. Therefore, for therapeutic approach, EMT TFs and signaling pathways should be targeted for curbing the genesis of CSCs through dedifferentiation.
Cell plasticity and CSC
Previously, the progression from stem cell to a more differentiated cell was considered to be unidirectional. CSC plasticity and bidirectional conversion between non-SC and SC concepts, however, have further complicated these postulations and should be able to explain the tumor heterogeneity (Figure 3). The transfection of adult cells with Oct4 [88], Klf4 [89], Sox2 [90] and c-Myc [91], which are termed as the Yamanaka factors, produces iPSCs [92]. Along with Nanog [93] they are substrates of the 26S eukaryotic proteasome and thus are spared from degradation in proteasome-deficient CSCs. Pluripotent transcription factor Oct4, expression of which is an important predictor of adverse clinical outcome in solid cancers [94,95] is involved in early embryonic development by interacting with Nanog locus [96,97]. Another transcription factor Kruppel-like factor 4 (Klf4) is crucial for the reprogramming of adult cells into pluripotency. Along with c-Myc, Klf4 can be replaced by LIN28 and Nanog [98]. Sox2 is a HMG-box transcription factor that forms hetero-dimers along with Oct4 and is also crucial for maintaining pluripotency in embryonic SCs [99]. It also controls self-renewal through the expression of nestin [100]. c-Myc oncogene belongs to the family of basic h-l-h leucine zipper transcription factors and acts as an universal enhancer of the expression of genes in SCs that greatly increases the competence of the other Yamanaka factors [101]. Recent reports from our laboratory have demonstrated that depending on the microenvironmental cues non-stem cancer cells and CSCs are interconvertible [52] thereby supporting the plastic nature of all cancer cells.
All the above factors have been implicated as key players in malignant transformation. Our ability to comprehend, the role of cellular plasticity on the function of tissues and disease formation, continues to evolve. Initially it was understood that in most somatic tissues, an adult cell performing a specialized function has restricted potential and is unable to produce a different type of cell. However, recent reports [102] concluded that there is a remarkable flexibility that allows the interconversion between SCs and non-SCs and vice versa, even going against the biochemical energy gradient. The notion that inherent plasticity is retained by terminally differentiated cells enhances the opportunities to target them in case of disease and thus restore normal function.
Transdifferentiation
Different patterns of neovascularization are exhibited by solid tumors including vasculogenesis, sprouting angiogenesis, transdifferentiation and vascular mimicry of CSCs [103]. The process by which tumor cells get incorporated into blood vessels to form a vascular structure that is similar to normal vessels is called vascular mimicry [104,105]. Vascular mimicry was first identified in melanoma wherein some cells co-expressed CSC and EC markers thereby being able to form vascular network [106]. This intermediate state is achieved by a phenomenon called transdifferentiation through CSCs directly contribute to the presence of ECs in tumor vessels (Figure 3). Since then, transdifferentiation has been discovered in a number of tumors, including breast, ovarian and lung cancers, GBM and sarcomas. GSCs transdifferentiate into Endothelial-Like Cells (ELC) independent of the cell fusion mechanism [107] that aid in tumor angiogenesis [108,109] CD105+ renal CSCs can generate ELCs in vitro and give rise to vessels with a human origin in vivo [110]. Human bCSCs also transdifferentiate into functional ELCs expressing endothelial markers like CD31, CD34 VE-Cadherin (CD144) in vitro and in vivo [111]. CD44+ ovarian CSCs transdifferentiate into ELCs in Matrigel and form CD34+blood vessels in xenograft tumor models [112]. Reports suggested that a large population of the ECs in glioblastoma has the same chromosomal aberrations as tumor cells. Transdifferentiation of CD133+ GSCs was further validated in a xenograft model in which ECs of human origin was detected without cell fusion. Furthermore, specific targeting of GSC-derived ECs abated tumor growth [109]. Wang et al. [108] discovered that CD105+ECs in GBM samples have GBM genetic mutations, indicating that these ECs are not derived from normal ECs [108]. Transdifferentiation of CD133+ GSCs is via endothelial progenitor cell status via Notch signaling pathway and not by nuclear fusion [108]. This VEGF-independent transdifferentiation is enhanced by the induction of HIF-1a [113]. These studies indicate that CSCs in multiple tumors have the ability to convert into ELCs to directly contribute to tumor angiogenesis. These results also partially explain the failure of anti-VEGF therapy since transdifferentiation of CSCs is VEGF-independent.
Drug Resistance
Chemo-therapy emerges as one of the most widespread used techniques but it is ineffective towards CSCs and results into relapse of cancer. CSCs escape anticancer therapies by the aid of the following mechanisms: (i) removal of therapeutic agents by drug efflux pumps, (ii) increased DNA damage repair pathways, (iii) activation of mitogenic/anti-apoptotic pathways, and (iv) ability to evade immune response [114].
ABC (ATP Binding Cassette) transporters is a superfamily consists of ABCC1, ABCB1 and ABCG2, enacting as multidrug efflux pumps at the expenditure of ATP. It has been seen in a study that the knockout ABC transporter gene model in mice demonstrates vulnerability to drugs [115]. ALDH1, an important marker of CSCs is also implicated in drug resistance through its chemical property of oxidizing aldehydes to carboxylic acids. Multiple ALDH1 inhibited models demonstrate reduced drug resistance capacity [116,117]. BCL-2, an anti-apoptotic protein, is thought to be involved in chemoresistance as it is observed to be upregulated in drug resistant CSC subpopulations. Targeting pathways renders the cells to be chemosensitive along with decreased levels of BCL-2 indicating direct relationship between drug resistance and levels of BCL-2 [116]. Wnt-β catenin signaling is undoubtedly related with CSC formation and drug resistance, hence, blocking of this pathway by siRNA causes the CSCs to be sensitized towards drugs, for example, CD133+ colon cancer cells became predisposed to 5-fluorouracil after abrogating Wnt cascade [118,119]. Notch cascade is tied-up with MDR associated proteins 1 as blocking notch via γ-secretase inhibitors or shRNA brings down the expression of these drug resistant proteins [120]. Additionally, NF-κB has been linked to paclitaxel and carboplatin resistance and blocking of this pathway results into apoptosis of drug resistant CSCs [116]. DNA damage response is also noted to cause chemo-resistance. CHK1, a component of this response is important for damage repair and cell cycle arrest. Inhibition of CHK1 makes the CSCs subject to the effect of drugs such as, gemcitabine in case of pancreatic tumor [121]. Dodging of immune surveillance by either downregulated expression of MHC-I, CD8+ or initiating apoptosis in immune effect or cells, enlists as few instances of evading rejection in ABCB5+ melanoma CSCs [122].
All these evidences suggest that CSCs are the main culprit behind drug resistance and therefore it becomes imperative to overcome this in order to reduce their influence on the overall progression of the disease. As a result, strategies for eradication and control of them are fundamental and are discussed below.
Therapy
Conventional therapeutic modalities of cancer comprise of surgery, radio-therapy and chemo-therapy which are usually used in combination of one another because combinatorial treatment strategies offer better therapy prospective. In adjuvant therapy, surgery is followed by chemo- or radiotherapy to help reduce the risk of cancer recurrence, while during neoadjuvant therapy chemo- or radiotherapy is given before surgery. Conventional chemotherapy is the use of chemicals or drugs, e.g., alkylating agents like hydrazines, plant alkaloids like taxanes, antitumor antibiotics like anthracyclines, anti-metabolites like purine antagonists and topoisomerase inhibitors, for treating cancer. In radio-therapy, radiation or specifically high energy rays like x-rays and electrons are used. However, both the modalities have their side effects because of generalized targeting of cancer cells as well as normal cells located in the vicinity, resulting in serious systemic and local toxicities which may even cause death in patients [123,124]. Moreover, these therapeutic techniques are not successful against all kinds of cancer and relapse of cancer is quite prevalent even after combinational therapy.
Drug resistance, recurrence of tumor and failure of radiation therapy can all be tracked down to the presence of CSCs [125]. CSCs are chemo-resistant and can give rise to new tumors even after surgery via its self-renewal, differentiation and metastatic properties. Hence, it becomes cardinal for targeting CSCs through novel procedures and use conventional techniques as a secondary mechanism to kill rest of the cancer cells. It is quite evident from the discussion above that targeting CSCs may become the next generation therapy of cancer. In recent years, many novel strategies have been devised to specifically target CSCs and their niche. In this regard, use of antibodies conjugated with cytotoxic agents or nanoparticles are found out to be effective [123,124]. Polymeric nanoparticles loaded with paclitaxel against CD133, a cell surface glycoprotein is expressed specifically on CSCs in solid tumors, decrease the cell number and colonies formed in colorectal adenocarcinoma as well as in the xenograft mouse model [126]. In lung cancer, targeting CTLA4, PD-1 and its ligand PD-L1 shows promising and reliable results [127]. A human monoclonal antibody, Ipilimumab, inhibits the binding of CTLA-4 to its corresponding ligand. A phase III study compares the effects of etoposide with those of a combination of etoposide and ipilimumab in heightening T cell responses and enhancing overall survival [128]. Also, in another ongoing phase III study, the combination of ipilimumab and nivolumab, which are human monoclonal antibodies and binds to PD-1, is being evaluated in small cell lung carcinoma patients [129].
Signaling pathways also serve as targets for CSC eradication. In fact, signaling cascades followed by normal stem cells are deregulated in CSCs. Therefore, to specifically inhibit CSCs without affecting the normal SCs is very crucial and so, small molecule inhibitors in nanoformulations are used along with chemo-therapeutic drugs [74]. In this respect, Notch pathway inhibitors like γ-secretase inhibitor and monoclonal antibodies are showing a promising future in cancer treatment [124]. Hedgehog and mTOR inhibitors such as vismodegib and rapamycin, respectively, provide an excellent solution against proliferation of CSCs and ALDH+ cells by down-regulating Nanog and Oct-4 levels [124,130]. Wnt/β catenin signaling can be obstructed by small molecule inhibitors and biological antagonists such as monoclonal antibodies and siRNA [124]. Nuclear transfer inhibitor, PP, of β catenin, also contributes in impeding the proliferation in lung adenocarcinoma CSCs [124,131]. Blocking of NF-κB by triptolide inhibits EMT transition [132]. Plant polyphenol curcumin has been found to inhibit breast CSC migration by augmenting the E-cadherin/β-catenin negative feedback loop [40]. Another report documents Mithramycin A-induced sensitization of breast CSC toward genotoxic drug doxorubicin [133]. Re-purposing of drugs is a new-age technology for targeting CSCs. Recently, anti-inflammatory drug Aspirin has been implicated in suppressing the acquisition of drug resistance in breast CSCs by dysregulating NF-κB-IL6 signaling [82].
Altering the microenvironment plays a crucial role in checking tumorigenesis. Drugs like Plerixafor, CTCE-9908 and NOX-A12 induce disjunction of the cancer cells from the stromal niche thereby making it more susceptible to drugs [123,124]. Such effect of these drugs also raises their possibility of being effective against CSCs. Niche also includes VEGF, responsible for angiogenesis and can be counteracted with mAb Bevacizumab [123]. Inhibitors of ABC transport pumps like MS-209 and tariquidar also provide a possibility for sensitizing CSCs [123]. Other auxiliary therapeutic modalities include silencing of oncogenic miRNAs, induction of CSC differentiation and apoptosis with the help of small molecule drugs [125]. Antisense oligonucleotide inhibition can target oncogenic miRNAs. In fact, knockdown of miR-21, which is frequently overexpressed in different CSCs [134], has been found to inhibit cell proliferation, metastasis and tumor growth in case of breast [135,136], ovarian [137], and lung cancers [138].
Conclusion
Over the past few decades, extensive research has led to the evolution of the cancer stem cell model. This has dramatically altered our ability to comprehend the underlying mechanisms of cancer growth and development. CSCs are intricately involved in the various stages of cancer progression starting from tumor initiation, angiogenesis, invasion, migration and drug resistance and they have the capability to differentiate into multiple lineages, further aiding the intra-tumoral heterogeneity. Also, CSCs maintain their pool having the potential to self-renew. Therefore, our discussion emphasizes on the varied aspects of cancer progression governed primarily by the CSCs, which has led to the portrayal of cancer as a stem cell disease. Our discussion also portrays that in the definition of CSCs, the phenomenon of plasticity, which permits the bidirectional transformation of non-stem cancer cells to CSCs and vice-versa, should be included. All these together support the notion defining cancer as a stem cell disease. Hence, the ultimate objective would be to target these drug-resistant CSCs for the successful regression of cancer. This would require the intervention of novel therapeutic procedures which would help overcome the shortcomings of the conventional therapies.
References
- Cancer. World Health Organization.
- Zakaria N, Yusoff N, Zakaria Z, Lim M, Baharuddin P, Fakiruddin K, et al. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer. 2015; 15: 84.
- Hanahan D, Weinberg R. Hallmarks of Cancer: The Next Generation. Cell. 2011; 144: 646-674.
- Kreso A, Dick J. Evolution of the Cancer Stem Cell Model. Cell Stem Cell. 2014; 14: 275-291.
- Visvader J, Lindeman G. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nature Reviews Cancer. 2008; 8: 755-768.
- Clarke M, Dick J, Dirks P, Eaves C, Jamieson C, Jones D, et al. Cancer Stem Cells--Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Research. 2006; 66: 9339-9344.
- Virchow R. Cellular-Pathologie. Archiv für Pathologische Anatomie und Physiologie und für Klinische Medicin. 1855; 8: 3-39.
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994; 367: 645-648.
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414: 105-111.
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003; 63: 5821-5828.
- Al-Hajj M, Wicha M, Benito-Hernandez A, Morrison S, Clarke M. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences. 2003; 100: 3983-3988.
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death and Differentiation. 2007; 15: 504-514.
- Ho M, Ng A, Lam S, Hung J. Side Population in Human Lung Cancer Cell Lines and Tumors Is Enriched with Stem-like Cancer Cells. Cancer Research. 2007; 67: 4827-4833.
- O’Brien C, Pollett A, Gallinger S, Dick J. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2006; 445: 106-110.
- Dalerba P, Dylla S, Park I, Liu R, Wang X, Cho R, et al. Phenotypic characterization of human colorectal cancer stem cells. Proceedings of the National Academy of Sciences. 2007; 104: 10158-10163.
- Yeung T, Gandhi S, Wilding J, Muschel R, Bodmer W. Cancer stem cells from colorectal cancer-derived cell lines. Proceedings of the National Academy of Sciences. 2010; 107: 3722-3727.
- Li C, Heidt D, Dalerba P, Burant C, Zhang L, Adsay V, et al. Identification of Pancreatic Cancer Stem Cells. Cancer Research. 2007; 67: 1030-1037.
- Simeone D. Pancreatic Cancer Stem Cells: Implications for the Treatment of Pancreatic Cancer. Clinical Cancer Research. 2008; 14: 5646-5648.
- Haygood C. Ovarian cancer stem cells: Can targeted therapy lead to improved progression-free survival?. World Journal of Stem Cells. 2014; 6: 441.
- Fang D. A Tumorigenic Subpopulation with Stem Cell Properties in Melanomas. Cancer Research. 2005; 65: 9328-9337.
- Valent P, Bonnet D, Wohrer S, Andreeff M, Copland M, Chomienne C, et al. Heterogeneity of Neoplastic Stem Cells: Theoretical, Functional, and Clinical Implications. Cancer Research. 2013; 3: 1037-1045.
- Tysnes B, Jerkvig R. Cancer initiation and progression: Involvement of stem cells and the microenvironment. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2007; 1775: 283-297.
- Sajithlal G, Rothermund K, Zhang F, Dabbs D, Latimer J, Grant S, et al. Permanently Blocked Stem Cells Derived From Breast Cancer Cell Lines. Stem Cells. 2010; 28: 1008-1018.
- Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014; 511: 246-250.
- Kim S, Kwon C, Nakano I. Detoxification of oxidative stress in glioma stem cells: Mechanism, clinical relevance, and therapeutic development. Journal of Neuroscience Research. 2014; 92: 1419-1424.
- Hsu H, Lin J, Hsu T, Su K, Wang C, Yang K, et al. Mesenchymal stem cells enhance lung cancer initiation through activation of IL-6/JAK2/STAT3 pathway. Lung Cancer. 2012; 75: 167-177.
- Niu G, Chen X. Vascular Endothelial Growth Factor as an Anti-Angiogenic Target for Cancer Therapy. Current Drug Targets. 2010; 11: 1000-1017.
- Zhu T, Costello M, Talsma C, Flack C, Crowley J, Hamm L, et al. Endothelial Cells Create a Stem Cell Niche in Glioblastoma by Providing NOTCH Ligands That Nurture Self-Renewal of Cancer Stem-Like Cells. Cancer Research. 2011; 71: 6061-6072.
- Ping Y, Bian X. Concise Review: Contribution of Cancer Stem Cells to Neovascularization. Stem Cells. 2011; 29: 888-894.
- Conley S, Wicha M. Antiangiogenic agents. Cell Cycle. 2012; 11: 1265-1266.
- Mimeault M, Batra S. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J Cell Mol Med. 2013; 17: 30-54.
- Sun X. Endothelial precursor cells promote angiogenesis in hepatocellular carcinoma. World Journal of Gastroenterology. 2012; 18: 4925.
- Zhao Y, Bao Q, Renner A, Camaj P, Eichhorn M, Ischenko I, et al. Cancer stem cells and angiogenesis. Int J Dev Biol. 2011; 55: 477-482.
- Shao ES, Lin L, Yao Y, Bostrom KI. Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood. 2009; 114: 197-206.
- Piccirillo S, Reynolds B, Zanetti N, Lamorte G, Binda E, Broggi G, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006; 444: 761-765.
- Hovinga K, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, et al. Inhibition of Notch Signaling in Glioblastoma Targets Cancer Stem Cells via an Endothelial Cell Intermediate. Stem Cells. 2010; 28: 1019-1029.
- Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer and Metastasis Reviews. 2009; 28: 15-33.
- Li Y, Wang L, Pappan L, Galliher-Beckley A, Shi J. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Molecular Cancer. 2012; 11: 87.
- Voon D, Wang H, Koo J, Chai J, Hor Y, Tan T, et al. EMT-Induced Stemness and Tumorigenicity Are Fueled by the EGFR/Ras Pathway. PLoS One. 2013; 8: 70427.
- Mukherjee S, Mazumdar M, Chakraborty S, Manna A, Saha S, Khan P, et al. Curcumin inhibits breast cancer stem cell migration by amplifying the E-cadherin/β-catenin negative feedback loop. Stem Cell Research & Therapy. 2014; 5: 116.
- Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends in Cell Biology. 2011; 21: 736-744.
- Gialeli C, Theocharis A, Karamanos N. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS Journal. 2010; 278: 16-27.
- Berges R, Tchoghandjian A, Honore S, Esteve M, Figarella-Branger D, Bachmann F, et al. The novel tubulin-binding, checkpoint activator BAL101553 inhibits EB1-dependent migration and invasion and promotes differentiation of glioblastoma stem-like cells. Molecular Cancer Therapeutics. 2016; 15: 2740-2749.
- Sanvoranart T, Supokawej A, Kheolamai P, U-pratya Y, Poungvarin N, Sathornsumetee S, et al. Targeting Netrin-1 in glioblastoma stem-like cells inhibits growth, invasion, and angiogenesis. Tumor Biology. 2016; 37: 14949-14960.
- Yokota J. Tumor progression and metastasis. Carcinogenesis. 2000; 21: 497-503.
- Vanharanta S, Massagué J. Origins of Metastatic Traits. Cancer Cell. 2013; 24: 410-421.
- Velasco-Velázquez M, Popov V, Lisanti M, Pestell R. The Role of Breast Cancer Stem Cells in Metastasis and Therapeutic Implications. The American Journal of Pathology. 2011; 179: 2-11.
- Brown L, Berse B, Van de Water L, Papadopoulos-Sergiou A, Perruzzi C, Manseau E, et al. Expression and distribution of osteopontin in human tissues: widespread association with luminal epithelial surfaces. Molecular Biology of the Cell. 1992; 3: 1169-1180.
- Subraman V, Thiyagarajan M, Malathi N, Rajan ST. OPN -Revisited. J Clin Diagn Res. 2015; 9: 3-10.
- Long H, Xie R, Xiang T, Zhao Z, Lin S, Liang Z, et al. Autocrine CCL5 Signaling Promotes Invasion and Migration of CD133+Ovarian Cancer Stem-Like Cells via NF-κB-Mediated MMP-9 Upregulation. Stem Cells. 2012; 30: 2309-2319.
- Kang Y, Siegel P, Shu W, Drobnjak M, Kakonen S, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003; 3: 537-549.
- Mukherjee S, Manna A, Bhattacharjee P, Mazumdar M, Saha S, Chakraborty S, et al. Non-migratory tumorigenic intrinsic cancer stem cells ensure breast cancer metastasis by generation of CXCR4+ migrating cancer stem cells. Oncogene. 2016; 35: 4937-494 8.
- Dick J. Stem cell concepts renew cancer research. Blood. 2008; 112: 4793-4807.
- Shackleton M, Quintana E, Fearon E, Morrison S. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell. 2009; 138: 822-829.
- Bonnet , Dick J. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Medicine. 1997; 3: 730-737.
- Meacham C, Morrison S. Tumour heterogeneity and cancer cell plasticity. Nature. 2013; 501: 328-337.
- Collins A. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Research. 2005; 65: 10946-10951.
- Curley M, Therrien V, Cummings C, Sergent P, Koulouris C, Friel A, et al. CD133 Expression Defines a Tumor Initiating Cell Population in Primary Human Ovarian Cancer. Stem Cells. 2009; 27: 875-883.
- Vermeulen L, Todaro M, de Sousa Mello F, Sprick M, Kemper K, Perez Alea M, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proceedings of the National Academy of Sciences. 2008; 105: 13427-13432.
- Suva M, Riggi N, Stehle J, Baumer K, Tercier S, Joseph J, et al. Identification of Cancer Stem Cells in Ewing's Sarcoma. Cancer Research. 2009; 69: 1776-1781.
- Huang Z, Wu T, Liu AY, Ouyang G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget. 2015; 6: 39550-39563.
- van Es J, van Gijn M, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/?-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005; 435: 959-963.
- Liu H, Zhang W, Jia Y, Yu Q, Grau G, Peng L, et al. Single-cell clones of liver cancer stem cells have the potential of differentiating into different types of tumor cells. Cell Death and Disease. 2013; 4: 857.
- Li Z, Rich J. Hypoxia and Hypoxia Inducible Factors in Cancer Stem Cell Maintenance. Current Topics in Microbiology and Immunology. 2010; 345: 21-30.
- Yan K, Wu Q, Yan D, Lee C, Rahim N, Tritschler I, et al. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes & Development. 2014; 28: 1085-1100.
- Sato T, van Es J, Snippert H, Stange D, Vries R, van den Born M, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2010; 469: 415-418.
- Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong J, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nature Cell Biology. 2010; 12: 468-476.
- Plaks V, Kong N, Werb Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells. Cell Stem Cell. 2015; 16: 225-238.
- Bao B, Ahmad A, Azmi A, Ali S, Sarkar F. Overview of Cancer Stem Cells (CSCs) and Mechanisms of Their Regulation: Implications for Cancer Therapy. Current Protocols in Pharmacology. 2013.
- Al-Hajj M, Clarke M. Self-renewal and solid tumor stem cells. Oncogene. 2004; 23: 7274-7282.
- Liu S, Dontu G, Wicha M. Breast Cancer Research. 2005; 7: 86.
- Xie J, Johnson RL, Zhang X, Bare JW, Waldman FM, Cogen PH, et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. 1997; 57: 2369-2372.
- Liu S. Hedgehog Signaling and Bmi-1 Regulate Self-renewal of Normal and Malignant Human Mammary Stem Cells. Cancer Research. 2006; 66: 6063-6071.
- Borah A, Raveendran S, Rochani A, Maekawa T, Kumar D. Targeting self-renewal pathways in cancer stem cells: clinical implications for cancer therapy. Oncogenesis. 2015; 4: 177.
- Friedmann-Morvinski D, Verma I. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Reports. 2014; 15: 244-253.
- Mani S, Guo W, Liao M, Eaton E, Ayyanan A, Zhou A, et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell. 2008; 133: 704-715.
- Gupta P, Onder T, Jiang G, Tao K, Kuperwasser C, Weinberg R, et al. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell. 2009; 138: 645-659.
- Kong D, Li Y, Wang Z, Sarkar F. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins. Cancers. 2011; 3: 716-729.
- Friedmann-Morvinski D, Bushong E, Ke E, Soda Y, Marumoto T, Singer O, et al. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science. 2012; 338: 1080-1084.
- Schwitalla S, Fingerle A, Cammareri P, Nebelsiek T, Göktuna S, Ziegler P, et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell. 2013; 152: 25-38.
- Bachoo R, Maher E, Ligon K, Sharpless N, Chan S, You M, et al. Epidermal growth factor receptor and Ink4a/Arf. Cancer Cell. 2002; 1: 269-277.
- Saha S, Mukherjee S, Khan P, Kajal K, Mazumdar M, Manna A, et al. Aspirin Suppresses the Acquisition of Chemoresistance in Breast Cancer by Disrupting an NFκB–IL6 Signaling Axis Responsible for the Generation of Cancer Stem Cells. Cancer Research. 2016; 76: 2000-2012.
- Scheel C, Weinberg R. Cancer stem cells and epithelial–mesenchymal transition: Concepts and molecular links. Seminars in Cancer Biology. 2012; 22: 396-403.
- Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008; 9: 82-589.
- Shimono Y, Zabala M, Cho R, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c Links Breast Cancer Stem Cells with Normal Stem Cells. Journal of End-to-End-testing. 2009; 138: 592-603.
- Guerra C, Schuhmacher A, Caamero M, Grippo P, Verdaguer L, Pérez-Gallego L, et al. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell. 2007; 11: 291-302.
- Yang M, Wu M, Chiou S, Chen P, Chang S, Liu C, et al. Direct regulation of TWIST by HIF-1a promotes metastasis. Nature Cell Biology. 2008; 10: 295-305.
- Xu H, Wang W, Li C, Yu H, Yang A, Wang B, et al. WWP2 promotes degradation of transcription factor OCT4 in human embryonic stem cells. Cell Research. 2009; 19: 796-796.
- Chen Z. Destabilization of Kruppel-Like Factor 4 Protein in Response to Serum Stimulation Involves the Ubiquitin-Proteasome Pathway. Cancer Research. 2005; 65: 10394-10400.
- Baltus G, Kowalski M, Zhai H, Tutter A, Quinn D, Wall D, et al. Acetylation of Sox2 Induces its Nuclear Export in Embryonic Stem Cells. Stem Cells. 2009; 27: 2175-2184.
- Farrell A, Sears R. MYC Degradation. Cold Spring Harbor Perspectives in Medicine. 2014; 4: 14365-14365.
- Yamanaka S, Blau H. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010; 465: 704-712.
- Moretto-Zita M, Jin H, Shen Z, Zhao T, Briggs S, Xu Y. Phosphorylation stabilizes Nanog by promoting its interaction with Pin1. Proceedings of the National Academy of Sciences. 2010; 107: 13312-13317.
- Saigusa S, Tanaka K, Toiyama Y, Yokoe T, Okugawa Y, Ioue Y, et al. Correlation of CD133, OCT4, and SOX2 in Rectal Cancer and Their Association with Distant Recurrence After Chemoradiotherapy. Annals of Surgical Oncology. 2009; 16: 3488-3498.
- Koukourakis M, Giatromanolaki A, Tsakmaki V, Danielidis V, Sivridis E. Cancer stem cell phenotype relates to radio-chemotherapy outcome in locally advanced squamous cell head–neck cancer. British Journal of Cancer. 2012; 106: 846-853.
- Levasseur D, Wang J, Dorschner M, Stamatoyannopoulos J, Orkin S. Oct4 dependence of chromatin structure within the extended Nanog locus in ES cells. Genes & Development. 2008; 22: 575-580.
- Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, et al. Formation of Pluripotent Stem Cells in the Mammalian Embryo Depends on the POU Transcription Factor Oct4. Cell. 1998; 95: 379-391.
- Yu J, Vodyanik M, Smuga-Otto K, Antosiewicz-Bourget J, Frane J, Tian S, et al. Induced Pluripotent Stem Cell Lines Derived From Human Somatic Cells. Obstetrical & Gynecological Survey. 2008; 63: 154-155.
- Boyer L, Lee T, Cole M, Johnstone S, Levine S, Zucker J, et al. Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell. 2005; 122: 947-956.
- Park D, Xiang A, Mao F, Zhang L, Di C, Liu X, et al. Nestin Is Required for the Proper Self-Renewal of Neural Stem Cells. Stem Cells. 2010; 28: 2162-2171.
- Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006; 126: 663-676.
- Cohen D, Melton D. Turning straw into gold: directing cell fate for regenerative medicine. Nature Reviews Genetics. 2011; 12: 243-252.
- Jain R, Carmeliet P. SnapShot: Tumor Angiogenesis. Cell. 2012; 149: 1408-1408.
- Wagenblast E, Soto M, Gutiérrez-ángel S, Hartl C, Gable A, Maceli A, et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature. 2015; 520: 358-362.
- Weis S, Cheresh D. Tumor angiogenesis: molecular pathways and therapeutic targets. Nature Medicine. 2011; 17: 1359-1370.
- Maniotis A, Folberg R, Hess A, Seftor E, Gardner L, Pe'er J, et al. Vascular Channel Formation by Human Melanoma Cells in vivo and in vitro: Vasculogenic Mimicry. The American Journal of Pathology. 1999; 155: 739-752.
- Wurmser A, Nakashima K, Summers R, Toni N, D'Amour K, Lie D, et al. Cell fusion-independent differentiation of neural stem cells to the endothelial lineage. Nature. 2004; 430: 350-356.
- Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga K, Geber A, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010; 468: 829-833.
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2011; 469: 432-432.
- Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. Identification of a tumor-initiating stem cell population in human renal carcinomas. The FASEB Journal. 2008; 22: 3696-3705.
- Bussolati B, Grange C, Sapino A, Camussi G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. Journal of Cellular and Molecular Medicine. 2008; 13: 309-319.
- Alvero A, Fu H, Holmberg J, Visintin I, Mor L, Marquina C, et al. Stem-Like Ovarian Cancer Cells Can Serve as Tumor Vascular Progenitors. Stem Cells. 2009; 27: 2405-2413.
- Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proceedings of the National Academy of Sciences. 2011; 108: 4274-4280.
- Rosa R, D’Amato V, Placido S, Bianco R. Approaches for targeting cancer stem cells drug resistance. Expert Opinion on Drug Discovery. 2016.
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature Reviews Cancer. 2005; 5: 275-284.
- Abdullah L, Chow E. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013; 2: 3.
- Bae I. Aldehyde dehydrogenase 1A1 confers intrinsic and acquired resistance to gemcitabine in human pancreatic adenocarcinoma MIA PaCa-2 cells. International Journal of Oncology. 2012; 41: 855-861.
- Deng Y, Pu X, Huang M, Xiao J, Zhou J, Lin T, et al. 5-Fluorouracil upregulates the activity of Wnt signaling pathway in CD133-positive colon cancer stem-like cells. Chinese Journal of Cancer. 2010; 29: 810-815.
- Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine. 2012; 7: 597-615.
- Cho S, Lu M, He X, Ee P, Bhat U, Schneider E, et al. Notch1 regulates the expression of the multidrug resistance gene ABCC1/MRP1 in cultured cancer cells. Proceedings of the National Academy of Sciences. 2011; 108: 20778-20783.
- Venkatesha V, Parsels L, Parsels J, Zhao L, Zabludoff S, Simeone D, et al. Sensitization of Pancreatic Cancer Stem Cells to Gemcitabine by Chk1 Inhibition. Neoplasia. 2012; 14: 519-525.
- Schatton T, Frank M. Antitumor Immunity and Cancer Stem Cells. Annals of the New York Academy of Sciences. 2009; 1176: 154-169.
- Chen K, Huang Y, Chen J. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacologica Sinica. 2013; 34: 732-740.
- Dragu D, Necula L, Bleotu C, Diaconu C, Chivu-Economescu M. Therapies targeting cancer stem cells: current trends and future challenges. World J Stem Cells. 2015; 7: 1185-1201.
- Han L, Shi S, Gong T, Zhang Z, Sun X. Cancer stem cells: therapeutic implications and perspectives in cancer therapy. Acta Pharmaceutica Sinica B. 2013; 3: 65-75.
- Swaminathan SK, Roger E, Toti U, Niu L, Ohlfest JR, Panyam J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J Control Release. 2013; 171: 280-287.
- Karachaliou N, Cao MG, Teixidó C, Viteri S, Morales-Espinoza D, Santarpia M, et al. Understanding the function and dysfunction of the immune system in lung cancer: the role of immune checkpoints. Cancer Biol Med 2015; 12: 79-86.
- Massarelli E, Papadimitrakopoulou V, Welsh J, Tang C, Tsao AS. Immunotherapy in lung cancer. Transl Lung Cancer Res 2014; 3: 53-63.
- Pillai RN, Owonikoko TK. Small cell lung cancer: therapies and targets. Semin Oncol 2014; 41: 133-142.
- Zuo M, Rashid A, Churi C, Vauthey J, Chang P, Li Y, et al. Novel therapeutic strategy targeting the Hedgehog signalling and mTOR pathways in biliary tract cancer. British Journal of Cancer. 2015; 112: 1042-1051.
- Zhang X, Lou Y, Zheng X, Wang H, Su J, Dong Q, et al. Wnt blockers inhibit the proliferation of lung cancer stem cells. DDDT. 2015; 9: 2399-2407.
- Liu L, Salnikov A, Bauer N, Aleksandrowicz E, Labsch S, Nwaeburu C, et al. Triptolide reverses hypoxia-induced epithelial-mesenchymal transition and stem-like features in pancreatic cancer by NF-κB downregulation. International Journal of Cancer. 2013; 134: 2489-2503.
- Saha S, Mukherjee S, Mazumdar M, Manna A, Khan P, Adhikary A, et al. Mithramycin A sensitizes therapy-resistant breast cancer stem cells toward genotoxic drug doxorubicin. Translational Research. 2015; 165: 558-577.
- Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J Biol Chem. 2007; 282: 14328-14336.
- Yan LX, Wu QN, Zhang Y, Li YY, Liao DZ, Hou JH, et al. Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivo tumor growth. Breast Cancer Res. 2011; 13: R2.
- Song B, Wang C, Liu J, Wang X, Lv L, Wei L, et al. MicroRNA-21 regulates breast cancer invasion partly by targeting tissue inhibitor of metalloproteinase 3 expression. J Exp Clin Cancer Res. 2010; 29: 29.
- Lou Y, Yang X, Wang F, Cui Z, Huang Y. MicroRNA-21 promotes the cell proliferation, invasion and migration abilities in ovarian epithelial carcinomas through inhibiting the expression of PTEN protein. Int J Mol Med. 2010; 26: 819-827.
- Shi SJ, Zhong ZR, Liu J, Zhang ZR, Sun X, Gong T. Solid lipid nanoparticles loaded with Anti-Microrna Oligonucleotides (AMOs) for suppression of microRNA-21 functions in human lung cancer cells. Pharm Res. 2012; 29: 97-109.