Case Report
Austin Surg Case Rep. 2021; 6(1): 1043.
Multiple Primary Intracranial and Spinal Juvenile Xanthogranuloma: A Case Report
Almanea AK1*, Alqazlan MS2, Bardisi MM3, Alotaibi F4 and Alshakweer W5
1Department of Pathology and Laboratory Medicine, King Abdulaziz Medical City, Saudi Arabia
2Department of Pathology and Laboratory Medicine, King Faisal Specialist Hospital and Research Center, Saudi Arabia
3Department of Pathology and Laboratory Medicine, Riyadh Regional Lab and Blood Bank, Ministry of Health, Saudi Arabia.
4Neuroscience Center, King Fahad Medical City, Saudi Arabia
5Department of Pathology and Laboratory Medicine, King Fahad Medical City, Saudi Arabia
*Corresponding author: Abdullah Khaled Almanea, Department of Pathology and Laboratory Medicine, King Abdulaziz Medical City, Saudi Arabia
Received: June 11, 2021; Accepted: July 31, 2021; Published: August 07, 2021
Abstract
Juvenile Xanthogranuloma (JXG) is a rare histiocytic disorder that belongs to the non-Langerhans cell histiocytosis family. It commonly occurs in the skin of young children, particularly the head and neck region. Occasional cases with extracutaneous involvement have been described. However, involvement of the central nervous system without cutaneous lesions is extremely rare. We present a case of an 11-year-old male child with multiple intracranial and spinal JXGs. At 30 months follow up, after administration of chemotherapy, the patient had passed away. The broad clinical spectrum of JXG and the morphological resemblance to other histiocytic lesions prompt a cautious approach for the diagnosis. Immunohistochemically, those lesions were positive for CD68 and negative for S100 and CD1a. A revised classification for histiocytosis was recently proposed, based on the underlying molecular characteristics. The diagnosis of extracutaneous or disseminated JXG with MAPK-activating mutation or ALK translocations was considered as Erdheim-Chester disease. However, our study did not fit into the proposed classification due to the absence of BRAF-V600E gene mutation and ALK gene rearrangement. Chemotherapy with or without radiotherapy has been suggested as treatment options for unresectable central nervous system lesions.
Keywords: JXG; Juvenile Xanthogranuloma; Histiocytic; Primary intracranial; Central nervus system histocytosis
Abbreviations
JXG: Juvenile Xanthogranuloma; CNS: Cntral Nervous System; MRI: Magnetic Resonance Imaging; PET/CT: Positron Emission Tomography Computed Tomography; GFAP: Glial Fibrillary Acidic Protein; AFP: Acid-Fast Bacilli; PAS: Periodic Acid-Schiff; GMS: Grocott’s Methenamine Silver Stain; RDD: Rosai-Dorfman Disease; LCH: Langerhans Cell Histiocytosis; ALK: Anaplastic Lymphoma Kinase; ECD: Erdheim-Chester Disease; MAPK/ERK: Mitogen- Activated Protein Kinase/Extracellular Signal-Regulated Kinases
Introduction
Juvenile Xanthogranuloma (JXG) is a benign cutaneous disorder of non-Langerhans cell histiocytic proliferation, which usually a selflimited disorder that occurs in children with a predilection for the head and neck region. It has been first described in 1905 and 1912 by Adamson and McDonagh, respectively [1,3]. It is the most common form of non-Langerhans cell histiocytosis. However, the true etiology of this disease is currently unknown [2]. JXG can be defined by the clinical setting as solitary to disseminated lesions, the areas of the body involved, and the patient’s age [3]. That is, It usually presents in the first year of life with one, several, or even numerous red to yellow cutaneous nodules that typically involve the head and neck area. These nodules usually range in size between 0.5 to 1.0 cm in diameter and are often self-limited. Extracutaneous dissemination is infrequent, primarily affecting the uveal tract of the eye [3]. Nevertheless, isolated lesion involving the Central Nervous System (CNS) without cutaneous involvement is extremely rare [4]. We are presenting a case of an 11-year-old female who presented with multiple primary intracranial and spinal JXGs without cutaneous manifestations.
Case Presentation
An 11-years old male child presented with long-standing right ear pain and erythema for two months that progressed into a decreased hearing with right facial nerve weakness and headache. Despite medical therapy, the patient had developed severe headaches and, subsequently tonic-clonic seizures. Physical examination showed right facial paralysis. All other sensory and motor functions were intact. No cutaneous lesions or bone tenderness were observed. Brain Magnetic Resonance Imaging (MRI) revealed multiple extra-axial and intraaxial supratentorial moderately enhancing lesions, including dural based, leptomeningeal based, and frontal region (Figure 1A,1B). In addition, bilateral vestibular lesions were seen in the internal auditory canals and the right trigeminal nerve (Figure 1C,1D). The largest lesion was dural-based and was located in the right temporal lobe. Spinal MRI exhibited multiple moderately enhancing spinal cord lesions at the level of C7, along the middle and lower thoracic, at the conus medullaris, the cauda equina, and the lower part of the theca’ sac at the level of S1 and S2 (Figure 1E,1F). No sclerotic or lytic bone lesions were identified on Positron Emission Tomography Computed Tomography (PET/CT). A chest, abdomen, and pelvis CT scans were negative for any organ involvements. A craniotomy approach with a dural-based lesional biopsy showed an inflammatory mass composed predominantly of histiocytes with scattered lymphocytes and occasional eosinophils. There were scattered clusters of Toutontype giant cells (Figure 2A). The tissue was negative for Glial Fibrillary Acidic Protein (GFAP) immunohistochemical stain excluding glial neoplasms. Additional staining with Acid-fast bacilli (AFB), Periodic Acid-Schiff (PAS), and Grocott’s Methenamine Silver (GMS) stain were also negative, excluding tuberculosis and infections causes. Positive immunohistochemical reaction for CD68 confirmed the histiocytic nature of the lesion, while negative reaction for S100 and CD1a excluded Rosai-Dorfman Disease (RDD) and Langerhans Cell Histiocytosis (LCH), respectively (Figure 2B-2D). Molecular study for BRAFV600E mutation and immunohistochemical stain for Anaplastic Lymphoma Kinase (ALK) were both negative, confirming the diagnosis of JXG and excluding Erdheim-Chester Disease (ECD) and ALK-positive histiocytosis, respectively (Figure 2E). The patient’s condition progressed despite chemo-radiotherapy, and he passed away 30 months from the time of initial diagnosis.
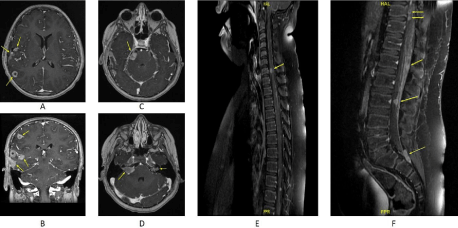
Figure 1: Brain and spinal Magnetic Resonance Imaging (MRI) on post-contrast T1-weighted sequence showing multiple supratentorial moderately enhancing
lesions (A, B). Additional enhancing lesions in the right trigeminal nerve (C) and bilateral vestibular lesions in the internal auditory canals (D). Spinal MRI show
multiple moderately enhancing lesions at the levels of C7 (E), middle and lower thoracic, conus medullaris, the cauda equina, and the lower part of the theca’ sac
at the level of S1 and S2 (F).

Figure 2: Hematoxylin-Eosin (H&E) section (A) demonstrates an inflammatory mass which consist of histiocytes and inflammatory cells in the form of lymphocytes
with clusters of Touton-type giant cells. Immunohistochemical stains shows positive reaction for CD68(B) with negative reaction for CD1a(C), S100(D), and ALK(E).
Discussion
The atypical presentation of the patient described in this report prompts a cautious approach to a xanthomatous lesion in the CNS. A wide variety of neoplastic and non-neoplastic diseases were considered in the initial differential diagnosis. For example, xanthomatous changes can frequently occur in meningioma and schwannoma [5]. It can also be seen in gliomas such as pleomorphic xanthoastrocytoma or other primary tumors such as craniopharyngioma and hemangioblastoma [5]. However, the morphological appearance and immunohistochemical results in our case exclude those tumors.
Infectious granulomatous conditions, such as extrapulmonary tuberculosis and fungal infections can appear morphologically similar and must be taken into consideration, especially the given widespread nature of the clinical course of JXG in this case. This differential is particularly relevant in areas where such infections are endemic. However, GMS and AFB special stains were negative for microorganisms.
Histiocytic neoplasms such as Rosai-Dorfman Disease, Langerhans cell histiocytosis, Erdheim Chester Disease, ALK-positive histiocytosis, and JXG are among the differential diagnosis in this case. The subtle morphological differences and immunohistochemistry profiles within the appropriate clinical history are critical in differentiating such cases.
Histologically, LCH is characterized by grooved nuclei (coffee bean appearance) and numerous eosinophils. RDD typically demonstrates histiocytes with central nuclei and engulfed lymphocytes (emperipolesis). JXG is characterized by foamy histiocytes and numerous Touton giant cells with an exuberant inflammatory background consisting of lymphoplasmacytic infiltrate as well as eosinophils and some neutrophils. Similarly, foamy histiocytes and Touton giant cells are present in ECD, but the inflammatory infiltrates are sparse in a more fibrotic background [6,7].
A panel of immunohistochemical antibodies including CD68, S100, CD1a, and ALK are required to reach a definitive diagnosis. CD68 and CD163 are general histiocytic antibodies seen in all inflammatory and neoplastic histiocytes including JXG and ECD. These two conditions can only be differentiated on a molecular basis; BRAFV600E mutation occurs only in ECD. S100 positivity in addition to CD163 with existing emperipolesis confirms the diagnosis of RDD. CD1a positivity combined with S100 and CD163 in the presence of coffee beans grooved histiocytes should be enough evidence to label the lesion LCH. However, Langerin is a rather Langerhans histiocytosis-specific marker. ALK-positive histiocytosis is a relatively recently described entity with different treatment protocol can be easily diagnosed by positive ALK immunohistochemical staining in the absence of other markers including SMA and CD45. Further confirmation can be done by the presence of the novel Kinesin Family Member 5B (KIF5B-ALK) gene fusion identified by Change et al [8].
Nowadays, however, with the advancing molecular classification including molecular testing for BRAF gene and Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinases (MAPK/ERK) pathway and Anaplastic Lymphoma Kinase (ALK) gene mutation give rise to more accurate differentiation between these lesions [9].
Recently, a revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages was proposed by the histiocyte society of blood. JXG with or without major systemic components is listed in the Non-Langerhans cell histiocytosis of skin and mucosa (C) group. On the other hand, extracutaneous or disseminated JXG with MAPK-activating mutation or ALK translocations are included within the Langerhans (L) group [9]. Our case did not fit in any of the proposed groupings by histiocyte society of blood given the absence of cutaneous lesions and the lack of ALK alteration in the immunohistochemical stains. MAPK/ERK genetic alterations testing was not available [9].
The recommended treatment option for solitary JXG is surgery as a first line of management. Unresectable lesions, particularly intracranial and disseminated primary CNS ones are treated with adjuvant chemotherapy alone or combined chemo-radiotherapy [10].
Conclusion
JXG is a rare mucocutaneous histiocytic proliferation that can disseminate to extracutaneous sites including CNS. However, a primary CNS disease without cutaneous manifestation is extremely rare, as in our case. JXG is one of the challenging cases to diagnose histopathologically because it needs a series of immunohistochemical and molecular testing.
References
- Fölster-Holst R. Severe systemic juvenile xanthogranuloma is an indication for systemic therapy. British Journal of Dermatology. 2017; 176: 302-304.
- Collie J, Harper C, Fillman E. Juvenile Xanthogranuloma. StatPearls Publishing. 2021.
- Dehner L. Juvenile Xanthogranulomas in the First Two Decades of Life: A Clinicopathologic Study of 174 Cases with Cutaneous and Extracutaneous Manifestations. The American Journal of Surgical Pathology. 2003; 27: 579- 593.
- De Paula A, André N, Fernandez C, Coulibaly B, Scavarda D, Lena G, et al. Solitary, extracutaneous, skull-based juvenile xanthogranuloma. Pediatric Blood & Cancer. 2010; 55: 380-382.
- Boström J, Janßen G, Messing-Jünger M, Felsberg J, Neuen-Jacob E, Engelbrecht V, et al. Multiple intracranial juvenile xanthogranulomas. Journal of Neurosurgery. 2000; 93: 335-341.
- Goyal G, Young J, Koster M, Tobin W, Vassallo R, Ryu J, et al. The Mayo Clinic Histiocytosis Working Group Consensus Statement for the Diagnosis and Evaluation of Adult Patients with Histiocytic Neoplasms: Erdheim-Chester Disease, Langerhans Cell Histiocytosis, and Rosai-Dorfman Disease. Mayo Clinic Proceedings. 2019; 94: 2054-2071.
- Hernández-San Martín M, Vargas-Mora P, Aranibar L. Juvenile Xanthogranuloma: An Entity with a Wide Clinical Spectrum. Actas Dermo- Sifiliográficas (English Edition). 2020; 111: 725-733.
- Chang K, Tay A, Kuick C, Chen H, Algar E, Taubenheim N, et al. ALK-positive histiocytosis: an expanded clinicopathologic spectrum and frequent presence of KIF5B-ALK fusion. Modern Pathology. 2018; 32: 598-608.
- Emile J, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016; 127: 2672-2681.
- Chen W, Cheng Y, Zhou S, Chen Y, Chen X, Xia S. Juvenile xanthogranuloma of central nervous system: Imaging of two cases report and literature review. Radiology of Infectious Diseases. 2017; 4: 117-120.