Research Article
Austin J Surg. 2014;1(7): 1034.
A Review of Management and Outcome of Colorectal Angiosarcoma
Dhebri AR1*, Chadwick M1, Chowdhury J2 and Loganathan S1
1Department of Surgery, Royal Albert Edward Infirmary, UK
2Department of Pathology, Salford Royal NHS Foundation Trust, UK
*Corresponding author: AR Dhebri, Department of Surgery, Royal Albert Edward Infirmary, Wigan Lane, Wigan, WN1 2NN, UK
Received: August 30, 2014; Accepted: September 22, 2014; Published: October 07, 2014
Abstract
Purpose: Colorectal angiosarcoma (AS) is a rare aggressive tumour, with no known natural history. We aim to determine the clinical course, management and prognosis of colorectal AS by an analysis of reported cases.
Methods: The cases were identified from PubMed, bibliographies of the retrieved papers and included one patient from our hospital.
Results: A total of 38 cases, including our one case, were analysed. The median (range) age at presentation was 59 (16-85) years and 55% were women. The most frequent site was sigmoid colon (39%). The size of the tumour ranged from 0.3-12 cms. (median: 5 cms.). The commonest symptom was rectal bleeding in 53%. Metastases were present in 48%, excluding the four cases which itself were metastases from AS at other sites. Surgical resection was carried out in 81%; chemotherapy given in 26% and radiotherapy in 28%. The median and 5-year survival rates were eight months and 2.9% respectively. On univariate analysis age, size of tumour, tumour ≥ 5 cm., recurrence and presence of metastases were significant but surgical resection including R0 resection, radiotherapy (XRT), chemotherapy and gender were not significant for survival. On multivariate analysis only the presence of metastases was significant.
Conclusion: AS has a poor prognosis compared to other colorectal tumours. The review suggests treatment of choice to be complete resection (R0), though because of very few cases published, there is no definite evidence for it. One must be aware of this rare entity, particularly in patient with colorectal malignancy presenting with unusual symptoms.
Keywords: Angiosarcoma; Sarcoma; Colon; Colorectal tumours
Introduction
Angiosarcoma (AS) is a rare soft tissue tumour accounting for less than 1% of all soft tissue sarcomas [1,2]. It rarely occurs in the Gastrointestinal Tract (GIT) [3,4], accounting for less than 1% of all GIT malignancies [5] and usually occurs in the stomach [6] and small intestine [4,6]. Primary colorectal AS are exceedingly rare, accounting for less than 0.001% of all colorectal cancers [3,7-9]. The first case of AS of the colon was described by Steiner et al in 1949 [10].
They can arise anywhere in the body but most commonly occur in the skin and superficial soft tissues of the head and neck (50 to 60%) [11,12], particularly in the scalp [11] of elderly white men. Rarely, intra-abdominal AS arises in the liver [13], spleen [14], ovary [15] and adrenal glands [16] or as diffuse angiosarcomatosis [17]. Metastases to the colon from AS elsewhere are exceedingly rare; the first case to be described was by Fujii et al. [18]. AS principally spreads haematogenously, with the lungs the most common site for metastases. Other sites for metastases include heart, pericardium, liver, spleen, adrenal glands, kidneys, lymph nodes, bones, brain and breast [11,19-25].
Because of the paucity of published cases, there is no overview of the clinical course and management of colorectal AS. The rarity of cases, difficulty in tissue diagnosis and the confusion of nomenclature in the past, may have caused a variety of cases to have been erroneously diagnosed (Table 1).
![]()
I)
Benign
Haemangiomas of subcut / deep soft tissue
capillary
cavernous
arteriovenous
venous
intramuscular
synovial
Epithelioid haemangioma
Angiomatosis
Lymphangioma
II)
Intermediate (locally aggressive)
Kaposiform haemangioendothelioma
III)
Intermediate (rarely metastasizing)
Retiform haemangioendothelioma
Papillary intralymphatic angioendothelioma
Composite haemangioendothelioma
Kaposi sarcoma
IV)
Malignant
Epithelioid haemangioendothelioma
Angiosarcoma of soft tissue
Table 1: WHO classification of Vascular Tumours.
Methods
Identification of patients of colorectal AS
AS is defined as a malignant aggressive tumour and develops from the endothelium of the lymphatics (lymphangiosarcoma) or blood vessels (haemangiosarcoma) [26]. We searched PubMed using the keywords angiosarcoma, sarcoma, colon, colorectal tumours, up to February 2013. Additional papers were identified from the bibliographies of the retrieved papers. We contacted the authors where possible for missing information and to enquire about the present status of the patients. Some of the early reports that were classified as endothelioma were included as there was invasion. Cases where the histology was unclear or where there was diffuse disease and site of origin was not clearly colorectal, were excluded. An additional case from our hospital was included.
Statistics
The data was described non-parametrically including median and range. The survival analysis was undertaken using Kaplan Meier and comparisons were analysed by Mann Whitney U test. The significance was set at p <0.05.
Results
Case report
A 48 years old woman, presented with anaemia, nausea, poor appetite, weight loss and no evidence of GIT bleeding. On examination, she had mild tenderness in left iliac fossa. A CT scan of abdomen showed a tumour in the proximal sigmoid colon with multiple liver and lung metastases and was staged as cT4 N1 M1. A colonoscopy confirmed a structuring lesion in the sigmoid colon. The biopsies from this lesion, done on two occasions were inconclusive. At operation, she was found to have a large locally advanced sigmoid tumour attached to the bladder. A left hemicolectomy was performed and the operation was considered palliative.
Post-operatively she had a pyrexia and tachycardia. A CT scan showed no anastigmatic or bladder leaks. However, it did show peripancreatic phlegmon and suspicious nodules in the anterior abdominal wall. She was commenced on chemotherapy but unfortunately she died of diffuse metastases, before completing the course in 2.5 months from surgery.
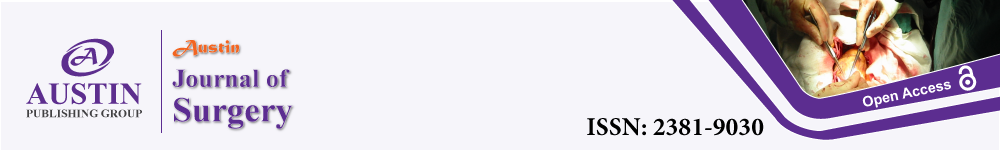
Histology showed an ulcerated annular tumour, measuring 12 cm, invading the full thickness of the bowel wall. Microscopy showed AS with variable vascular pattern, being poorly differentiated, with lots of rudimentary vascular channels, with spindle morphology. The vascular channels lined by atypical endothelial cells and scattered foci of tumour necrosis (Figure 1). There was positivity for endothelial markers CD31 and CD34 confirming the diagnosis. Though no definite bladder invasion could be identified, complete excision could not be confirmed. None of the 25 lymph nodes were positive for tumour.
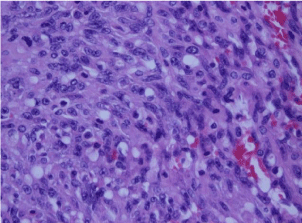
Figure 1:(x20 magnification) – H&E section showing poorly differentiated component at higher magnification, demonstrating the rudimentary vascular channels, atypical endothelial cells with pleomorphic nuclei, prominent mitoses and spindly morphology.
Literature review
In the literature, majority of the papers were either isolated case reports [3,9,10,17,18,25-48] or small retrospective GI case series containing some colorectal cases [6,49-51]. Two cases were from a collective review of GIT vascular tumours [52]. A total of 40 papers [3,6,7,9-11,14,17,18,25-55] were identified, of which five [7,14,53-55] were excluded as they did not meet the inclusion criteria. The PRISMA (Preferred Reporting Items for Systematic Reviews and Meta- Analyses) flow diagram (Figure 2) depicts the flow of information in this review. PRISMA is an evidence-based minimum set of items for reporting in systematic reviews and meta-analyses.
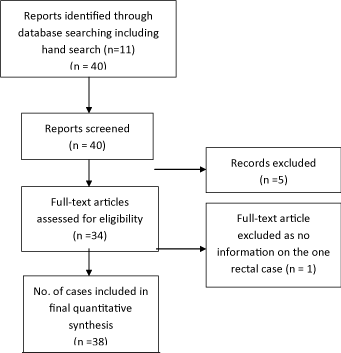
Figure 2: PRISMA Flow Diagram.
One more paper, a review of AS at all sites, with one rectal case was excluded as there was no information on the case at all [11]. In total, 38 cases (37 cases from 34 papers plus our case) were included for the analysis. In the present study we report on the natural history of the disease by detailed case report analysis {Figures in parenthesis are number of cases where information is available}. The largest review reported, in the form of a letter, consists of 23 cases of colorectal AS [45]. The three large reviews on AS at all sites [20,22,56], which did not mention any colorectal case, were also reviewed.
The age range {38} was 16 to 85 years (median: 59 years) with 21 females (55%) [6,10,17,26,31,32,35,37,39-41,43,47-49,50-52]. The commonest site {38} was sigmoid (15; 39 %) [3,9,10,19,25- 27,29,34,37,38,43,49,50]. Other sites were caesium, ascending colon, transverse colon, descending colon and rectum. In five cases, multiple sites were involved such as stomach, duodenum, small bowel, omentum, peritoneum and musculoskeletal [29,32,33,37,41]. The size of the tumour {28} ranged from 0.3-12 cms. (median 5 cms.). The size of tumour ≥ 5cms. was significant for survival (p=0.04) [6,18,28,29,31,36-38,40,43,44,46,48,52]. The causes {37} suggested were radiotherapy in three cases [17,28,46], Foreign Body (FB) in two [40,44], chronic anorectal ulceration in one [30] and possibly long-term dialysis in one case [33]. AS was primary in 32 cases (86%) and secondary in four (11%) [18,25,29,50], with metastasis from scalp, bone, breast and lungs in each.
The commonest symptom {36} was rectal bleeding in 19 (53%) [3,27,29,30,34-38,40,45-50,52]. Other symptoms were abdominal pain, abdominal mass, rectal mass, bowel obstruction, weight loss, anaemia, anal / perianal pain, melena and asymptomatic. One patient with AS of caecum presented with intussusception [42]. CT scan {25} was reported to be done in 13 (52%) [3,12,18,25,27-29,32,34,43,44,46] and endoscopic examination {27} in 21 (77%) [3,9,18,25,27-29,32- 37,40,43-46,48,49]. Preoperative endoscopic biopsy {26} was done in 17 (65%) [3,9,18,25,28,29,30,33-35,37,40,45,46,48,52] and was positive for malignant cells in ten (58%), with a definitive report of AS in four of these [18,30,33,45]. Metastases {37} were present in 18 (48%) [3,6,9,10,17,32-34,36,38-41,43,45,52], excluding the four cases which itself were metastases from AS at other sites [18,25,29,50]. The common sites of metastases were lungs in seven [9,25,32,33,50,52], liver in six [31,33,38,50] and bones in six [25,33,34,39,40,50]. Other sites of metastases were adrenal, duodenum, GIT, spleen, muscle / soft tissues and cervical lymph node.
Surgical resection {37} was carried out in 30 cases (81%) [3,6,9,10,17,18,26-32,34-40,42-46,48-50], with no surgery performed in one [52], endoluminal resection in three [25,47,52], duodenal excision in one [41] and gastroduodenectomy in one [33]. The last two cases had multifocal AS. The lymph nodes {17} were positive in seven (41%) [3,27,28,32,33,42,45]. Adjuvant treatment {35} was given in 16 (46%); chemotherapy {34} in nine (26%) [27,29,32,34,45,50] and radiotherapy {35} in ten (28%) [10,27,31,40,47,50,52]. The survival {34} ranged from 0.5 to 156 months (median 8 months) with 5-yr. survival of 2.9% (Figure 3) and 2-yr. survival 38%. Mortality {35} was 18 (53%) [3,6,9,17,28,31,33,36,38-41,43,44,50,52] and 14 were known to be alive without disease [10,26,27,29,30,35,37,42,45,47-50,52]. Recurrence {32} intra-abdominally or at other sites occurred in six (19%) [6,18,28,36,43,44]. On univariate analysis, age (p=0.03), any size of tumour (p=0.03), size of tumour ≥ 5cm. (p=0.04), recurrence (p=0.05) and metastases (p=0.0003) were significant but surgical resection including R0 resection, XRT, chemotherapy, age > 50 yrs. and gender were not significant for survival (Mann Whitney U test). On multivariate analysis only presence of metastases was significant (Table 2).
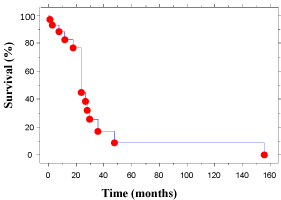
Figure 3: Meier Survival Curve for Colorectal Angiosarcoma.
![]()
Variable
Univariate (p value)
Multivariate (p value)
Age
p=0.03*
p=0.08
Age > 50 yrs
p=012
p=0.14
Gender
p=0.65
p=0.42
Any size of tumour
p=0.03*
p=0.11
Size of tumour ≥ 5cm
p=0.04*
p=0.16
Surgical resection
p=0.16
p=0.99
R0 resection
p=0.49
p=0.94
Radiotherapy
p=0.10
p=0.34
Chemotherapy
p=0.20
p=0.38
Recurrence
p=0.05*
p=0.42
Metastases
p=0.0003*
p=0.005*
Table 2: Univariate and Multivariate Analysis.
Discussion
Histology
Histologic diagnosis can be difficult, with variable features [49]. They are characterized by the formation of anastomosing, irregular, angulated vascular channels lined by abnormal, pleomorphic, malignant endothelial cells [6,18,50] or could be lined by large anaplastic cells [27]. In poorly differentiated areas, the malignant endothelial cells form continuous sheets, usually with an epithelioid morphology and with areas of haemorrhage and necrosis, which can make differentiation from anaplastic carcinoma difficult [1,3,7,19]. Deep seated AS frequently has an epithelioid morphology. The epithelioid phenotype of AS was first described in cases of cutaneous AS by Rosai et al. [57] in 1976.
Immunohistochemistry, although helpful, there is no consistently reliable test for AS. It is positive for some or all of the endothelial markers like CD31, CD34, factor VIII related antigen [58], von Wille brand factor and Vascular Endothelial Growth [19] Factor (VEGF). Von Wille brand factor, Ulex europaeus agglutinin I, and CD31 are the most useful markers in poorly differentiated cases [59]. However, tumour dedifferentiation can lead to a loss of these markers.
Epitheliod AS has characteristic features. CD31 and CD34 confirm the endothelial component, while epithelial markers like cytokeratin, classify it as epithelioid AS [58], leading to confusion with poorly differentiated carcinomas. Endothelin-1-like immunoreactivity seems to be a good marker for this type of AS [49]. No comprehensive studies of molecular changes in AS have been published. Gene-expression microarray technology should help to improve diagnosis, particularly in early or poorly differentiated cases and suggest potential therapeutic targets [19].
Differential diagnosis
The differential diagnosis includes gastrointestinal stromal tumour, Kaposi’s sarcoma, malignant melanoma, leiomyosarcoma and benign GI angiomatoses [49]. Epithelioid hemangioendothelioma, a vascular neoplasm with more indolent behaviour and a propensity for multifocal involvement, has a lobular architecture. In contrast, epithelioid AS, a variant of AS, tends to grow in diffuse sheets with larger more rounded pleomorphic ‘epithelioid’ endothelial cells with abundant eosinophilic cytoplasm containing prominent vesicular nucleoli and subtle clefting, indicative of vascular differentiation. These two neoplasms may represent a biologic continuum [50].
Aetiopathogenesis
AS at all sites, are believed to arise spontaneously [60], though some believe it may arise from benign vascular tumours or neurofibromas [19,61]. Although the causality has not been clearly elucidated, several factors have been incriminated in AS at all sites (Table 3). Vinyl chloride, arsenic, thorium dioxide [62], insecticides [63], radium, anabolic steroids and synthetic oestrogens [64,65] have been reported to cause AS in the liver [62]. Radiotherapy, chemotherapy, chronic inflammation, chronic ulceration, chronic lymphoedema (usually post mastectomy: Stewart-Treves syndrome), congenital lymphoedema (Milroy’s disease) or filarial lymphoedema [66-71] are important predisposing factors for AS. Rarely, it may develop in association with foreign materials, especially metals, dacron graft or a retained surgical sponge. In the literature, four cases of AS developing in the abdominal cavity in relation to a gauze retained for a prolonged period after previous abdominal surgery have been reported [40,54,72,73]. Radiation has been associated with usually skin and superficial tissues AS including breast, with few cases of post irradiation intestinal AS, though a definitive association is difficult to establish. The contribution of immunosuppression to the pathogenesis of AS is uncertain, though it has been reported in patients following renal transplant [74]. Familial syndromes including neurofibromatosis, Maffucci syndrome and Klippel- Trenaunay syndrome are also associated with AS [1].
![]()
• Radiation
- Chronic lymphoedema (Stewart-Treves syndrome)
Postsurgery or radiotherapy
Milroy's syndrome
Other types of chronic I ymphoedema
- Exogenous toxins
Vinyl chloride
Thorium dioxide
Arsenic
Anabolic steroids
Foreign bodies
- Familial syndromes
Neurofibromatosis NF-1
Mutated BRCA1 or BRCA2
Maffucci syndrome
Klippel-Trenaunay syndrome
Table 3: Risk factors for Angiosarcoma.Clinical signs
Clinical signs are similar to common colorectal cancers, usually presenting with anaemia, abdominal pain, abdominal mass, GI bleeding and bowel obstruction. In 2004, Brown et al. summarised 13 reported cases of colorectal AS in the literature [3]. Majority of the patients (61%) were female and age ranged from 16 to 77 years. The location was sigmoid (n=5) dominantly and most patients presented with rectal bleeding (n=7). In another paper on a series of GIT AS, the most frequently presenting symptom was also intestinal bleeding [50]. In some cases the presentation is because of intraperitoneal bleeding [26,36,43]. There can be secondary involvement of bowel from a primary somewhere as mentioned above or from direct spread from retro peritoneum. A metastatic involvement of the duodenum from primary in colon is a rarer finding at upper endoscopy causing GI bleeding [9]. Due to the diagnostic difficulties, metastases are already present in many patients at the time of diagnosis. In a paper by Abraham et al., on 82 patients with AS at all sites, it mentioned localised disease in 50–80% (our review 40%) and 20–45% (our review 59%) has metastatic disease at presentation [21].
Investigations
In non-emergency cases, diagnostic assessment includes CT scan of thorax, abdomen and pelvis and MRI pelvis (in rectal lesions), to delineate the extent of the primary lesion and metastases. CT reveals the heterogenous enhancement of the lesion, similar to that of AS in the liver [75] and spleen [76]. Colonoscopy and endoscopic biopsy, to confirm histology, is commonly performed, but may not confirm diagnosis. PET imaging may be useful to detect metastases and recurrence [77].
Treatment
There are no randomised trials, though there is some evidence from larger studies involving AS at all sites. Because of the rarity of AS, optimal management has not been well defined and the role of adjuvant treatment for colorectal AS is unclear.
Surgery: Radical surgery seems to be the primary treatment of choice and may be the only curative option. Complete (R0) resection should be performed where possible, with wide margins, though local and distant recurrences are common [11,19]. In the review by Brown et al. of colorectal AS [3], all survivors had a surgical resection of the tumour and none received adjuvant therapy and they concluded that surgical resection was the only curative treatment option. In our review, there was no significant correlation between surgical resection including R0 resection and survival. Involved margins are common because of in apparent spread beyond macroscopic margins conferring a worse prognosis [20,21,23,55].
Radiotherapy (XRT): Because of the high risk of local recurrence, adjuvant XRT is recommended by Mark et al. in a review of AS at all sites [11], though XRT alone was inadequate treatment for potentially curable disease. No formal XRT trials have been done, but retrospective series of AS at other sites, suggest that it improves local control and overall survival [23,78] although some investigators have failed to identify any benefit [21,55,56,79]. XRT is usually avoided for radiation-induced AS [19] because of maximum dose being reached [80].
Chemotherapy: The effect of chemotherapy on AS overall is reported as disappointing and there is no evidence that it prolongs overall survival. In the series by Mark et al. of AS at all sites, adjuvant chemotherapy did not significantly improve Disease Free Survival (DFS) [11]. In metastatic tumour dissemination, chemotherapy may be the primary treatment option, although the evidence is limited [19].
The chemotherapeutic agents used were doxorubicin, cyclophosphamide, methotrexate, vincristine, ifosfamide and more recently taxanes [11,19]. In AS of head and neck, breast and extremity demonstrated longer overall survival than did those with AS of visceral sites, suggesting biologic heterogeneity of AS [37,81].
Multimodal treatment: In a Japanese series by Naka et al. of 55 AS at all sites (including head and neck, extremities and organs), multimodal treatment (surgery and chemotherapy with or without XRT) was associated with a better prognosis [56]. In the series by Mark et al. of AS at all sites [11], showed that the addition of local radiotherapy resulted in improved DFS (43%), compared with surgery (13%) as the only local modality (p = 0.03).
In our review, we did not find multimodal treatment having any significant survival benefit.
Although the underlying pathogenic pathways are not fully understood, newer antiangiogenic drugs targeting VEGF pathways such as bevacizumab, sorafenib and sunitinib [19] have been tried. Trials of these drugs are showing promise [19].
Prognosis
Prognosis is poor with only rare cases with long-term survival. In the series by Mark et al., the overall 5-yr. actuarial survival was 35% and actuarial 5-yr. DFS was 24% [19]. Our review showed 5-yr. survival of 2.9%. The better survival in the series by Mark et al. is because majority of the cases in their series affected head, neck and extremities, which have better survival.
Brown et al., in their review on colorectal AS, found that age above 60 and size of tumour ≥ 5 cm. had poor prognosis [3]. Similarly Naka et al. [56] series of AS at all sites, showed on univariate analysis that age ≥ 50 years, size ≥ 5 cm., mitotic counts ≥10 / 10HPF and multimodal treatment had significantly better 2-year survival. A multivariate analysis in that series, suggested tumour size, mode of treatment and mitotic counts were independent prognostic factors. In our review, univariate analysis showed age (p=0.03), size of tumour (p=0.03), size of tumour ≥ 5cm. (p=0.04), recurrence (p=0.05) and metastases (p=0.0003) were significant but surgical resection including R0 resection, XRT, chemotherapy, age > 50 yrs. and gender were not significant for survival. On multivariate analysis only presence of metastases was significant.
There are limitations of the review as we have searched only one database and also the potential problem of basing a review solely on published case reports. The treatment and outcomes may have changed over the period covered by the literature review.
These cases may be best treated in a specialist centre.
Conclusion
Current evidence for management of colorectal AS is limited because of very few cases reported. Surgery seems to be the main curative treatment option with wide resection margins. One must be aware of this rare entity, particularly in a patient with unusual symptoms and attempt to achieve R0 resection. Despite a substantial risk of subsequent recurrence and metastatic disease there is no evidence for benefit of adjuvant chemotherapy or XRT. Palliative chemotherapy in metastatic disease may be reasonable. Our algorithm for management is suggested in Figure 4. Biological therapies need further prospective studies to clarify their role in treatment.
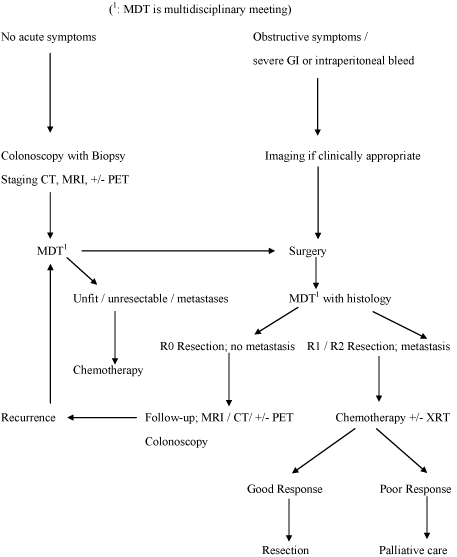 Figure 4:Suggested Algorithm for the Management of Angiosarcoma of Colon.Figure 4: Suggested Algorithm for the Management of Angiosarcoma of Colon.
Figure 4:Suggested Algorithm for the Management of Angiosarcoma of Colon.Figure 4: Suggested Algorithm for the Management of Angiosarcoma of Colon.References
- Enzinger FM, Weiss SW. Soft tissue tumors. St.Louis: CV Mosby Company. 1988: 545-580.
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 5th edn. St Louis, MO: Mosby. 2007.
- Brown CJ, Falck VG, MacLean A. Angiosarcoma of the colon and rectum: report of a case and review of the literature. Dis Colon Rectum. 2004; 47: 2202-2207.
- Chami TN, Ratner LE, Henneberry J, Smith DP, Hill G, Katz PO. Angiosarcoma of the small intestine: a case report and literature review. Am J Gastroenterol. 1994; 89: 797-800.
- Shaver TR, Lee YT. Nonosseous sarcomas in a military hospital. J Surg Oncol. 1987; 36: 284-289.
- Taxy JB, Battifora H. Angiosarcoma of the gastrointestinal tract. A report of three cases. Cancer. 1988; 62: 210-216.
- DiSario JA, Burt RW, Kendrick ML, McWhorter WP. Colorectal cancers of rare histologic types compared with adenocarcinomas. Dis Colon Rectum. 1994; 37: 1277-1280.
- Ryu DY, Hwang SY, Lee DW, Kim TO, Park DY, Kim GH, et al. [A case of primary angiosarcoma of small intestine presenting as recurrent gastrointestinal bleeding]. Korean J Gastroenterol. 2005; 46: 404-408.
- Cammarota G, Schinzari G, Larocca LM, Gasbarrini G, Barone C. Duodenal metastasis from a primary angiosarcoma of the colon. Gastrointest Endosc. 2006; 63: 330.
- Steiner CA, Palmer LH. Angiosarcoma of the Colon: With Case Report. Ann Surg. 1949; 129: 538-542.
- Mark RJ, Poen JC, Tran LM, Fu YS, Juillard GF. Angiosarcoma. A report of 67 patients and a review of the literature. Cancer. 1996; 77: 2400-2406.
- Yang JC, Rosenberg SA, Glatstein EJ, Antman KH. Sarcomas of soft tissue. 4th edn. DeVita VT, Hellman S, Rosenberg SA, editors. In: Cancer: principles and practice of oncology, Philadelphia: J.B. Lippincott. 1993: 1436-1455.
- Neshiwat LF, Friedland ML, Schorr-Lesnick B, Feldman S, Glucksman WJ, Russo RD. Hepatic angiosarcoma. Am J Med. 1992; 93: 219-222.
- Ghani M, Coughlin BF, Hickey KL, Reynolds DR. Angiosarcoma: unusual cause of acute abdominal pain and hemoperitoneum. Emergency Radiology. 2000; 7: 308-311.
- Nucci MR, Krausz T, Lifschitz-Mercer B, Chan JK, Fletcher CD. Angiosarcoma of the ovary: clinicopathologic and immunohistochemical analysis of four cases with a broad morphologic spectrum. Am J Surg Pathol. 1998; 22: 620-630.
- Wenig BM, Abbondanzo SL, Heffess CS. Epithelioid angiosarcoma of the adrenal glands. A clinicopathologic study of nine cases with a discussion of the implications of finding "epithelial-specific" markers. Am J Surg Pathol. 1994; 18: 62-73.
- Wolov RB, Sato N, Azumi N, Lack EE. Intra-abdominal "angiosarcomatosis" report of two cases after pelvic irradiation. Cancer. 1991; 67: 2275-2279.
- Fujii Y, Koibuchi-Yamaoka H, Taniguchi N, Yasuda Y, Nagai H. Metastasis from a primary angiosarcoma of the scalp to the colon: sonographic and CT findings. J Clin Ultrasound. 2008; 36: 110-112.
- Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. Lancet Oncol. 2010; 11: 983-991.
- Fury MG, Antonescu CR, Van Zee KJ, Brennan MF, Maki RG. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer J. 2005; 11: 241-247.
- Abraham JA, Hornicek FJ, Kaufman AM, Harmon DC, Springfield DS, Raskin KA, et al. Treatment and outcome of 82 patients with angiosarcoma. Ann Surg Oncol. 2007; 14: 1953-1967.
- Naka N, Ohsawa M, Tomita Y, Kanno H, Uchida A, Aozasa K. Angiosarcoma in Japan. A review of 99 cases. Cancer. 1995; 75: 989-996.
- Pawlik TM, Paulino AF, McGinn CJ, Baker LH, Cohen DS, Morris JS, et al. Cutaneous angiosarcoma of the scalp: a multidisciplinary approach. Cancer. 2003; 98: 1716-1726.
- Lahat G, Dhuka AR, Lahat S, Smith KD, Pollock RE, Hunt KK, et al. Outcome of locally recurrent and metastatic angiosarcoma. Ann Surg Oncol. 2009; 16: 2502-2509.
- Ricchetti T, Paci M, Cavazza A, Ferrari G, Annessi V, De Franco S, et al. A case of metastatic epithelioid angiosarcoma in the lamina propria of a sigmoid tubulovillous adenoma. Tumori. 2005; 91: 210-212.
- Lo TH, Tsai MS, Chen TA. Angiosarcoma of sigmoid colon with intraperitoneal bleeding: case report and literature review. Ann R Coll Surg Engl. 2011; 93: 91-93.
- Komorowski AL, Darasz Z, Koaodziejski LS, Gruchaa A. Primary angiosarcoma of the colon. Acta Chir Belg. 2004; 104: 465-467.
- Tardio JC, Najera L, Alemany I, Martin T, Castano A, Perez-Regadera JF. Rectal angiosarcoma after adjuvant chemoradiotherapy for adenocarcinoma of the rectum. J Clin Oncol. 2009; 27: 116-117.
- Sadhu S, Pattari S, Shaikh F, Verma R, Roy MK. Colonic metastasis from subcutaneous angiosarcoma: A diagnostic dilemma. Indian J Surg. 2010; 72: 328-330.
- Lo YM, Gillett MB, Vina M, Collin J, Fleming KA. Hemangiosarcoma of the rectum after chronic anorectal ulceration. J Clin Gastroenterol. 1989; 11: 77-81.
- ORMOS J, SIN L. [Hemangiosarcoma of the intestine]. Zentralbl Allg Pathol. 1959; 99: 352-357.
- Joseph UA, Jhingran SG. Technetium-99m labelled red blood cells in the evaluation of hemangiosarcoma. Clin Nucl Med. 1987; 12: 845-847.
- Usuda H, Naito M. Multicentric angiosarcoma of the gastrointestinal tract. Pathol Int. 1997; 47: 553-556.
- Al Beteddini OS, Brenez D, Firket C, Algaba R, Tabech A. Colonic angiosarcoma: A case report and review of literature. Int J Surg Case Rep. 2013; 4: 208-211.
- Pascual M, Juanpere N, Alameda F, Pera M. Primary colonic angiosarcoma as a cause of massive lower gastrointestinal bleeding. Rev Esp Enferm Dig. 2010; 102: 288-289.
- Saito R, Bedetti CD, Caines MJ, Kramer K. Malignant epithelioid hemangioendothelioma of the colon. Report of a case. Dis Colon Rectum. 1987; 30: 707-711.
- Smith JA, Bhathal PS, Cuthbertson AM. Angiosarcoma of the colon. Report of a case with long-term survival. Dis Colon Rectum. 1990; 33: 330-333.
- Hofman P, Bernard JL, Michiels JF, Saint Paul MC, Rampal A, Benchimol D, et al. [Primary angiosarcoma of the colon. Anatomo-clinical study of a case]. Ann Pathol. 1991; 11: 25-30.
- Solheim K, Dammen I, Roald BB, Carlsen E. [Angiosarcoma of the colon]. Tidsskr nor Laegeforen. 1991; 111: 3068-3069.
- Ben-Izhak O, Kerner H, Brenner B, Lichtig C. Angiosarcoma of the colon developing in a capsule of a foreign body. Report of a case with associated hemorrhagic diathesis. Am J Clin Pathol. 1992; 97: 416-420.
- Deléaval JP, Peter MY, Laurencet F, Fontolliet C. [Multicentric intestinal angiosarcoma. Report on a case]. Ann Pathol. 1991; 11: 342-344.
- Rozario A, Ravi HR. Angiosarcoma of cecum: unusual presentation with intussusceptions. Indian J Gastroenterol. 1995; 14: 31-32.
- El Chaar M, McQuay N. Sigmoid colon angiosarcoma with intraperitoneal bleeding and early metastasis. J Surg Educ. 2007; 64: 54-56.
- Joo YT, Jeong CY, Jung EJ, Lee YJ, Hong SC, Choi SK, et al. Intra-abdominal angiosarcoma developing in a capsule of a foreign body: report of a case with associated hemorrhagic diathesis. World J Surg Oncol. 2005; 3: 60.
- Hinterseher I, Pistorius S, Zietz C, Bergert H. Epithelioid hemangiosarcoma of the rectum. Int J Colorectal Dis. 2005; 20: 385-387.
- Ahmed I, Hamacher KL. Angiosarcoma in a chronically immunosuppressed renal transplant recipient: report of a case and review of the literature. Am J Dermatopathol. 2002; 24: 330-335.
- Morgan CN. Endothelioma of the Rectum. Proc R Soc Med. 1932; 25: 1020-1021.
- Norbury LE. Specimen of Endothelioma of Rectum. Proc R Soc Med. 1932; 25: 1021-1022.
- Watanabe K, Hoshi N, Suzuki T, Suzuki T. Epithelioid angiosarcoma of the intestinal tract with endothelin-1-like immunoreactivity. Virchows Arch A Pathol Anat Histopathol. 1993; 423: 309-314.
- Allison KH, Yoder BJ, Bronner MP, Goldblum JR, Rubin BP. Angiosarcoma involving the gastrointestinal tract: a series of primary and metastatic cases. Am J Surg Pathol. 2004; 28: 298-307.
- Alles JU, Bosslet K. Immunocytochemistry of angiosarcomas. A study of 19 cases with special emphasis on the applicability of endothelial cell specific markers to routinely prepared tissues. Am J Clin Pathol. 1988; 89: 463-471.
- Gentry RW, Dockerty MB, Glagett OT. Vascular malformations and vascular tumors of the gastrointestinal tract. Surg Gynecol Obstet. 1949; 88: 281-323.
- Pereira TC, Vi LH, Prichard JW, Sturgis CD. Pathologic quiz case. Elderly man with bright red blood per rectum. Pathologic diagnosis: metastatic hepatic epithelioid angiosarcoma. Arch Pathol Lab Med. 2001; 125: 968-970.
- Jennings TA, Peterson L, Axiotis CA, Friedlaender GE, Cooke RA, Rosai J. Angiosarcoma associated with foreign body material. A report of three cases. Cancer. 1988; 62: 2436-2444.
- Fayette J, Martin E, Piperno-Neumann S, Le Cesne A, Robert C, Bonvalot S, et al. Angiosarcomas, a heterogeneous group of sarcomas with specific behaviour depending on primary site: a retrospective study of 161 cases. Ann Oncol. 2007; 18: 2030-2036.
- Naka N, Ohsawa M, Tomita Y, Kanno H, Uchida A, Myoui A, et al. Prognostic factors in angiosarcoma: a multivariate analysis of 55 cases. J Surg Oncol. 1996; 61: 170-176.
- Rosai J, Sumner HW, Kostianovsky M, Perez-Mesa C. Angiosarcoma of the skin. A clinicopathologic and fine structural study. Hum Pathol. 1976; 7: 83-109.
- Delvaux V, Sciot R, Neuville B, Moerman P, Peeters M, Filez L, et al. Multifocal epithelioid angiosarcoma of the small intestine. Virchows Arch. 2000; 437: 90-94.
- Ohsawa M, Naka N, Tomita Y, Kawamori D, Kanno H, Aozasa K. Use of immunohistochemical procedures in diagnosing angiosarcoma. Evaluation of 98 cases. Cancer. 1995; 75: 2867-2874.
- Stout AP. Hemangio-Endothelioma: A tumor of blood vessels featuring vascular endothelial cells. Ann Surg. 1943; 118: 445-464.
- Rossi S, Fletcher CD. Angiosarcoma arising in hemangioma/vascular malformation: report of four cases and review of the literature. Am J Surg Pathol. 2002; 26: 1319-1329.
- Lee FI, Smith PM, Bennett B, Williams DM. Occupationally related angiosarcoma of the liver in the United Kingdom 1972-1994. Gut. 1996; 39: 312-318.
- Mazanet R, Antman KH. Sarcomas of soft tissue and bone. Cancer. 1991; 68: 463-473.
- Falk H, Thomas LB, Popper H, Ishak KG. Hepatic angiosarcoma associated with androgenic anabolic steroids. Lancet Hoch-Ligeti C. Angiosarcoma of liver associated with diethystilbesterol. JAMA. 1978; 240: 1510-1511.
- Hoch-Ligeti C. Angiosarcoma of the liver associated with diethylstilbestrol. JAMA. 1978; 240: 1510-1511.
- STEWART FW, TREVES N. Lymphangiosarcoma in postmastectomy lymphedema; a report of six cases in elephantiasis chirurgica. Cancer. 1948; 1: 64-81.
- Rosenberg WA. Hereditary edema of the legs (Milroy's disease). Archives of Dermatology. 1940; 42: 1113-1121.
- Muller R, Hajdu SI, Brennan MF. Lymphangiosarcoma associated with chronic filarial lymphedema. Cancer. 1987; 59: 179-183.
- Aitola P, Poutiainen A, Nordback I. Small-bowel angiosarcoma after pelvic irradiation: a report of two cases. Int J Colorectal Dis. 1999; 14: 308-310.
- Aozasa K, Naka N, Tomita Y, Ohsawa M, Kanno H, Uchida A, et al. Angiosarcoma developing from chronic pyothorax. Mod Pathol. 1994; 7: 906-911.
- Nanus DM, Kelsen D, Clark DG. Radiation-induced angiosarcoma. Cancer. 1987; 60: 777-779.
- Cokelaere K, Vanvuchelen J, Michielsen P, Sciot R. Epithelioid angiosarcoma of the splenic capsule. Report of a case reiterating the concept of inert foreign body tumorigenesis. Virchows Arch. 2001; 438: 398-403.
- Keymeulen K, Dillemans B. Epitheloid angiosarcoma of the splenic capsula as a result of foreign body tumorigenesis. A case report. Acta Chir Belg. 2004; 104: 217-220.
- Ahmed I, Hamacher KL. Angiosarcoma in a chronically immunosuppressed renal transplant recipient: report of a case and review of the literature. Am J Dermatopathol. 2002; 24: 330-335.
- Koyama T, Fletcher JG, Johnson CD, Kuo MS, Notohara K, Burgart LJ. Primary hepatic angiosarcoma: findings at CT and MR imaging. Radiology. 2002; 222: 667-673.
- Thompson WM, Levy AD, Aguilera NS, Gorospe L, Abbott RM. Angiosarcoma of the spleen: imaging characteristics in 12 patients. Radiology. 2005; 235: 106-115.
- Maeda T, Tateishi U, Hasegawa T, Ojima H, Arai Y, Sugimura K. Primary hepatic angiosarcoma on coregistered FDG PET and CT images. AJR Am J Roentgenol. 2007; 188: 1615-1617.
- Schlemmer M, Reichardt P, Verweij J, Hartmann JT, Judson I, Thyss A, et al. Paclitaxel in patients with advanced angiosarcomas of soft tissue: a retrospective study of the EORTC soft tissue and bone sarcoma group. Eur J Cancer. 2008; 44: 2433-2436.
- Sher T, Hennessy BT, Valero V, Broglio K, Woodward WA, Trent J, et al. Primary angiosarcomas of the breast. Cancer. 2007; 110: 173-178.
- Kuten A, Sapir D, Cohen Y, Haim N, Borovik R, Robinson E. Postirradiation soft tissue sarcoma occurring in breast cancer patients: report of seven cases and results of combination chemotherapy. J Surg Oncol. 1985; 28: 168-171.
- Janigan DT, Husain A, Robinson NA. Cardiac angiosarcomas. A review and a case report. Cancer. 1986; 57: 852-859.