Special Article - Surgical Case Reports
Austin J Surg. 2015;2(3): 1056.
Gallbladder Dysgenesis Requiring Reoperation for Cholecystectomy: A Case Report
Stephen Serio* and Maher Ghanem
Department of Surgery, Central Michigan University College of Medicine, USA
*Corresponding author: Stephen Serio, Department of Surgery, Central Michigan University College of Medicine, 901 S. Washington, Suite 1, Saginaw, MI 48603, USA
Received: March 09, 2015; Accepted: March 23, 2015; Published: March 26, 2015
Abstract
In this case report, we describe a unique case of gallbladder dysgenesis fraught with diagnostic dilemma and operative misadventure. We include a brief review of the literature regarding the pathophysiologic and embryologic etiology, as well as the incidence of this rare finding.
Introduction
Anatomic variation is a common and well-studied aspect of hepatobiliary surgery. Frequently, anomalies involving the biliary tree conformation and vascular supply are witnessed by hepatobiliary and general surgeons at operation, however, embryologic anomalies of the gallbladder are much less frequently encountered.
Embryologic irregularity of the gallbladder is a rare finding, occurring in less than 0.1% of the population [1]. The incidence of congenital absence of the gallbladder is even less frequent, occurring in about 0.03% [2]. These findings are rarely described in the literature, occasionally reported in reviews of necropsy and radiographic case studies. Less commonly, diagnosis is made intraoperatively; in the case surgery is under taken in light of false positive ultrasonography exams [3]. It is difficult to predict incidence from these limited reports.
Congenital absence and dysgenesis of the gallbladder was found to be asymptomatic in many adult cases, occasionally diagnosed at surgery for misinterpreted or misdiagnosed biliary complaints [6]. It appears as though these malformations occur randomly in the population, typically without a familial predisposition. This seems to indicate that these phenotypic changes are the result of sporadic mutations. Retrospectively, gallbladder agenesis is identified as an isolated defect in 70-82% of those affected. Of these, only 55.6% are found to be symptomatic [7]. Of note, agenesis has also been identified, incidentally, in conjunction with midgut volvulus and cholangiocarcinoma [7,8].
In autopsy studies, pediatric populations are more likely to have associated anatomic defects, which often limit lifespan. These include malformations most commonly of the genitourinary, reproductive, cardiovascular and gastrointestinal tract. Additionally, though less frequently, anomalies were identified in the respiratory tract and skeletal system [2]. Congenitally, multiple syndromes have been associated with agenesis of the gallbladder. These include cerebrotendinous xanthomatosis and Opitz’s syndrome, among others [9]. In addition to these generalized systemic conditions, hepatobiliary abnormalities, such as biliary atresia and congenital dysgenesis of the liver are also common concomitant findings. These conditions are sometimes not identified until later in life when the patient becomes symptomatic in their early adult years [10].
In this case report, we describe a unique case of gallbladder dysgenesis. Frequently, these variations are found posthumously, however our report examines the management and surgical treatment of gallbladder dysgenesis. At initial operation, what was presumed to be gallbladder agenesis, congenital absence of the gallbladder, was revealed by further symptomatic episodes and radiographic workup, and later at reoperation, to be dysgenesis of gallbladder formation, thus resulting in a small contracted gallbladder bud deep in the hepatic portal recess.
Case Report
A forty-one-year-old female was evaluated in our clinic with complaints of right upper quadrant and epigastric abdominal pain, worse in the postprandial setting and associated with some nausea and dyspepsia. She had no associated symptoms of fevers or chills. Her vital signs were unremarkable and physical exam reveals mild tenderness to deep palpation at the right sub costal margin.
The patient had previously undergone work up for similar complaints about three years prior to our evaluation. At that time, an ultrasound revealed an intrahepatic, contracted gallbladder with cholelithiasis. A minimally invasive surgery trained surgeon offered a laparoscopic cholecystectomy. At laparoscopy, the operating team was unable to identify a gallbladder and the cholecystectomy was aborted.
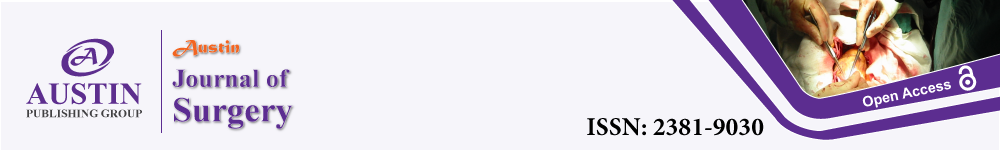
Subsequently, the patient underwent Magnetic Resonance Cholangiopancreatography (MRCP), which again revealed a contracted gallbladder with intraluminal stone and wall enhancement (Figure 1). Despite recommendation for reoperation, the patient had decided upon medical management and was lost to follow up for three years, at which time she was referred to our clinic for evaluation following an episode of presumed biliary colic.
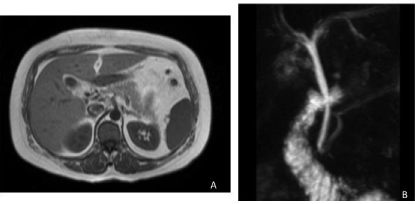
Figure 1a and 1b: Magnetic Resonance Cholangiopancreatography (MRCP)
performed after the initial operation, identifying a contracted gallbladder with
intraluminal stone and wall enhancement.
At surgery, the gallbladder fossa was examined. About 3cm from the liver edge, a contracted gallbladder without an obvious fund us was identified (Figure 2a). This was deep within the recess of the porta hepatitis and appeared to be in close relation to the portal structures. This was grasped, with difficulty, given the fact that the body was completely replaced with a large hard cholesterol gallstone. Dissection of the critical view of safety progressed from a lateral approach and the cystic duct was identified. A cholangiogram was performed confirming the anatomy of the biliary tree without any additional anomaly (Figure 3). Next, the duct and the artery were cleanly dissected and ligated and the malformed gallbladder was removed from the fossa (Figure 2b).
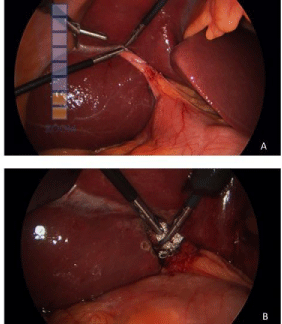
Figure 2a and 2b: The malformed gallbladder identified deep within the porta
hepatis prior to and following removal. The gallbladder is seen retracted and
without a fund us, consistent with dysgenesis of the gallbladder.
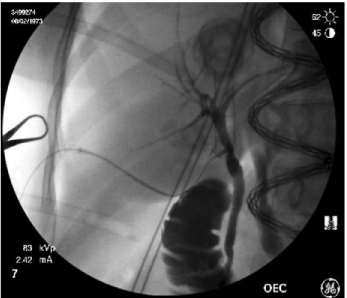
Figure 3: Intraoperative cholangiogram reveals a short cystic duct, nearly
abutting the common bile duct, though without filling defect and a normal
intrahepatic biliary tree.
Following the procedure, the patient recovered without incident. The histopathological evaluation of the specimen was consistent with inflamed gallbladder, consistent with our diagnosis of biliary colic.She was able to return to her routine diet and activity without issue or complication.
Discussion
Embryological development of the biliary tree begins during the fourth week of gestation as the hepatic diverticulum develops in the ventral wall of the midgut. As these structures become more defined, buds begin to develop into recognizable structures. By the fifth gestational week, all parts of the biliary tree are identifiable; with the gallbladder and cystic duct developing from caudal buddings of the initial diverticular out pouching [11].
The common duct develops distally and is initially a solid structure. Canalization of the lumen of the extrahepatic biliary tree begins near the seventh gestational week and is complete by the twelfth week when the gallbladder develops into a cystic structure. The liver and intrahepatic biliary tree develops from cephalad budding of the original hepatic diverticulum and these structures near complete maturation by the tenth gestational week. After a clockwise rotation around the duodenum, the ventral pancreatic bud and the extrahepatic biliary tree are in their final conformation and bile begins to flow into the duodenum by late in the twelfth gestational week [12].
Genetic transcription factors facilitate development of the biliary tree and the expression or the absence of these factors may account for the abnormalities seen in our patient. The intra and extrahepatic biliary tree appears to develop from distinct progenitor cells. Specifically, progenitor cells co expressing the SOX17 sequence and Pancreatic and Duodenal Homeobox Factor-1 (PDX1) develop into the extrahepatic ducts and pancreatic and duodenal structures. The absence of SOX17 and conversion to sole PDX1 expression appears to result in gallbladder agenesis, along with development of ectopic pancreatic tissues [13].
Additionally, other transcription factors appear to play a role in gallbladder dysgenesis and extrahepatic duct dysgenesis, as well. Hepatocyte nuclear factors, specifically HNF-6 and HNF1-β appear to play a direct role in the development and maturation of the gallbladder and common bile duct. Knockout mice lacking these specific factors were shown to lack a gallbladder and have abnormal development of the common bile duct or lack of canalization [14].
Both, agenesis and dysgenesis of the gallbladder are rare phenomena. This fact likely compounds the difficulty is diagnosing these anomalies, as traditional radiographic modalities have a high rate of false positive results, and most are accurately diagnosed only at operative exploration, as was the case for our patient [15]. Furthermore, a thorough understanding of the embryologic development and the possible variance in anatomy is critical for the practicing surgeon. In our case, this may have avoided the necessary reoperation and delay of definitive treatment.
References
- Peloponissios N, Gillet M, Cavin R, Halkic N. Agenesis of the gallbladder: a dangerously misdiagnosed malformation. World J Gastroenterol. 2005; 11: 6228-6231.
- Turkel SB, Swanson V, Chandrasoma P. Malformations associated with congenital absence of the gall bladder. J Med Genet. 1983; 20: 445-449.
- Haoues N, Zairi S, Zaafouri H, Ben Maamer A, Noomene R, Oueslati A, et al. Gallbladder agenesis intraoperatively diagnosed: a case report. Tunis Med. 2014; 92: 168-169.
- Kabiri H, Domingo OH, Tzarnas CD. Agenesis of the gallbladder. Curr Surg. 2006; 63: 104-106.
- Sherson ND. The absent adult gallbladder. Aust NZJ Surg. 1970; 39: 255-258.
- Fiaschetti V, Calabrese G, Viarani S, Bazzocchi G, Simonetti G. Gallbladder agenesis and cystic duct absence in an adult patient diagnosed by magnetic resonance cholangiography: Report of a case and review of the literature. Case Reports in Medicine. 2009; 674768.
- Yoldas O, Yazici P, Ozsan I, Karabuga T, Alpdogan O, Sahin E, et al. Coexistence of gallbladder agenesis and cholangiocarcinoma: report of a case. J Gastrointest Surg. 2014; 18: 1373-1376.
- Calder N, Carneiro HA, Khwaja HA, Thompson JN. Gallbladder agenesis with midgut malrotation. BMJ Case Rep. 2012.
- Majithia R, Salcedo JA. Absence of a gallbladder does not always mean a cholecystectomy. Gastrointest Endosc. 2012; 76: 1248-1249.
- Joliat GR, Shubert CR, Farley DR. Isolated congenital agenesis of the gallbladder and cystic duct: report of a case. J Surg Educ. 2013; 70: 117-120.
- Bedi N, Bond-Smith G, Kumar S, Hutchins R. Gallbladder agenesis with choledochal cyst--a rare association: a case report and review of possible genetic or embryological links. BMJ Case Rep. 2013.
- Ando H. Embryology of the biliary tract. Dig Surg. 2010; 27: 87-89.
- Academic Press. Development, Differentiation and Disease of the Para-Alimentary Tract. London, UK: Elsevier. 2010.
- Clotman F, Lannoy VJ, Reber M, Cereghini S, Cassiman D, Jacquemin P, et al. The onecut transcription factor HNF6 is required for normal development ofthe biliary tract. Development. 2002; 129: 1819-1828.
- Azmat N, Francis KR, Mandava N, Pizzi WF. Agenesis of the gallbladder revisited laparoscopically. Am J Gastroenterol. 1993; 88: 1269-1270.