Special Article – Acute Lung Injury
Austin J Surg. 2019; 6(1): 1159.
Acute Lung Injury Associated Milieus Regulate the Plasticity of Type I Alveolar Cells via KLF2
Yang X, Sun J, Liu D, Du J, Yang C, Wang H, Wen D, Zeng L* and Jiang J*
State Key Laboratory of Trauma, Burns and Combined Injury, Third Military Medical University, China
*Corresponding author: Jian-xin Jiang and Ling Zeng, Research Institute of Surgery, Daping Hospital, Third Military Medical University, China
Received: November 20, 2018; Accepted: January 07, 2019; Published: January 14, 2019
Abstract
Efficient repair and regeneration of alveolar epithelium after acute lung injuries are critical for pulmonary maintenance. Previous studies suggested alveolar type II cells (ATIIs) are progenitors in the adult lung and Alveolar Type I cells (ATIs) are terminally differentiated. However, recent studies develop fresh perspectives, which suggest ATIs may play more roles. Here we show that ATIs exhibit markedly enhanced proliferation capacity and express a set of ATIIs associated genes after treated with damaged lung milieus. These changed characteristics suggest ATIs generate phenotypic plasticity and exhibit dedifferentiated state. We identify KLF2 as a regulator of the module. Knockdown of KLF2 induces gene expression is consistent to those observed in milieu treated cells. These findings demonstrate the unanticipated plasticity of ATIs and suggest the molecular mechanisms controlling ATI-ATII plasticity which deserve more explorations.
Keywords: KLF2; Alveolar cells; ATI; ATII
Introduction
Adult lung consists of two major biologically distinct components including airway compartments and gas-exchanging units. Different component has region-specific stem/progenitor cells, which are critical for repair and regeneration following various injuries to rebuild functional structures. Mature alveoli are covered with two kinds of epithelial cells, type I and type II epithelial cells [1-6]. ATIs are thin cells covering 95-99% of the alveolar surface area, and lie closely with capillaries to facilitate gas diffusion. ATII cells, which cover the rest of the alveolar surface, are cuboidal cells and secrete surfactant proteins. Classical studies suggest ATIs are terminally differentiated, arise from ATIIs during alveolar homeostasis and regeneration process.
However, with advances in technology, fresh perspectives on the plasticity of ATIs are developed [7,8]. When cultured in vitro, ATIs exhibit some plasticity including the capacities to proliferate and express OCT-4, a protein that is involved in establishing and maintaining the undifferentiated pluripotent state. In vivo studies show us that part of ATIs can regenerate ATIIs in the lung and proliferate upon pneumonectomy or oncogenic KRAS expression. Nevertheless, it is still unclear whether ATIs can adopt new phenotypes to participate in regenerative responses? How do ATIs adopt their new phenotypes?
In this study, we focus on the poorly understood plasticity of ATIs in vitro. As alveolar milieus are capable of mobilizing different kinds of cells participating in repair through regulating their characteristics, we use lung extract from ARDS rats to mimic damaged milieu and explore how the plasticity of ATIs changed. To our surprise, ATIs exhibit markedly enhanced proliferation capacity and express a set of ATIIs associated genes after treated with damaged lung extract. These changed characteristics suggested ATIs generate phenotypic plasticity and exhibit dedifferentiated state. Then we explore the regulators of the module. We find KLF2 is contribute to the changed characteristics [9]. Knockdown of KLF2 induces gene expression changes similar to those observed in milieu treated cells. These findings demonstrate the unanticipated plasticity of ATIs and suggest the molecular mechanisms controlling ATI plasticity deserve more explorations, thus further establishing novel therapeutic approaches in treating lung diseases.
Materials and Methods
Animals
All the animals used here were male pathogen-free Sprague- Dawley rats providing by the Third Military Medical University animal center. These animals were housed in pathogen-free facility under a 12:12-h light-dark cycle with controlled room temperature (21-28°C) and humidity, and allowed ad libitum access to food and water. All animal studies were performed in accordance with the local Guide for the Care and Use of Laboratory Animals and were approved by the ethic committee.
Isolation and culture of ATIs
Rat ATIs were isolated and purified using a modification of published methods. Briefly, male SD rat (ag. 4-6-week-old; wt. 100- 160 g) lungs were dissected, perfused, lavaged, and then instilled with dispase (10 U/ml; Corning) for 50 min at 37°C. The resulting lungs were minced, filtered, washed, and incubated in rat IgG-coated Petri dishes in DMEM for 30 min at 37°C. Unattached cells were collected, resuspended in DMEM with 5% FBS, and incubated in dishes for 20 min at 37°C to loosen non-epithelial cells to the extent possible. Unattached cells were collected again, incubated with T1a antibody (5 mg/ml; sigma) in DMEM with 1% FBS for 45 min at 4°C on a rotator, washed and incubated with anti-rabbit IgG microbeads for 15 min at 4°C. The suspensions were washed and selected for ATIs. The purity of fresh isolated T1a-positive cells (AEC1s) was 86.6 ± 1.62%.
Freshly isolated ATIs were seeded at a density of 3×104 cells/ cm2 in high-glucose DMEM supplemented with 10% FBS, 100 units (mg)/ml of penicillin and streptomycin, and maintained in a 5% CO2 humidified 37°C incubator. Culture medium was refreshed every other day.
Acute lung injury animal model
Acute lung injury model was induced through Hemorrhagic Shock/Resuscitation (HR) combined with transtracheal injection of lipopolysaccharide (Escherichia coli serotype O55: B5, Difco Laboratories, Detroit, MI) as previously described. In short, 250±20 g male rats were anesthetization and shaved off the hair on interfeminium. Left femoral artery was carefully isolated, ligated and inserted a catheter to monitor of Mean Arterial Pressure (MAP), blood sampling, and resuscitation. Hemorrhagic shock was triggered by sucking blood, leading to the MAP to 40 mm Hg within 10 min. This blood pressure was maintained by further pumping blood for 1.5 hours. After that, rats were resuscitated with pumped blood plus equal volume of Ringer’s solution, and injected transtracheally 250 ul LPS (4.5 mg/kg in sterilized saline). The sham-operated animals were subjected to surgical procedures and injection of 250 ul sterilized saline without hemorrhagic shock/resuscitation and LPS challenge. All the works were operated cautiously to reduce infectious diseases or other undesirable outcomes.
Preparation of lung extract from normal and injured lungs
Rats were sacrificed at days 1, 3, 5 and 7 after transtracheal injection, respectively. Whole Lung was excised, perfused, segmented. Left lobes were fixed in 4% paraformaldehyde, dehydrated in ethanol, embedded in paraffin. Right lobes were transferred to the mortar containing liquid nitrogen and finely powdered. Then tissue powder was dissolved in DMEM, centrifuged, filtered, aliquoted and frozen at -80°C.
KLF2 silencing by small interfering RNA transfection in ATI cells
ATIs were transfected with validated small interfering RNA (siRNA) duplex oligonucleotides (Genepharma, Shanghai, China) by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) as the the transfection reagent according to the manufacturer’s instructions. In brief, ATI cells were grown routinely in DMEM with 10% fetal bovine serum to about 50% confluence, and then treated with 100 nM KLF2 siRNA, or a non-specific duplex siRNA as negative control using lipofectamine. Transfected cells were grown at 37°C for 4h, followed by incubation with fresh culture medium. After 24h or 48h, transfected cells were examined with phasecontrast microscopy, and used in further assays or RNA/protein extraction. The sequences for KLF2 siRNAs and control siRNA used here were: KLF2-rat-104 (sense: 5'-G C A C G G A U G A G G A C C U A A A T T-3', antisense: 5'-U U U A G G U C C U C A U C C G U G C T T-3'); the control siRNA 5 (sense: 5'-U U C U C C G A A C G U G U C A C G U T T-3', anti-sense: 5'-A C G U G A C A C G U U C G G A G A A T T-3').
CCK-8 assay
To monitor cell growth after the treatment of different factors, ATI cell proliferation was quantified by Cell Counting Kit-8 (CCK- 8) (Beyotime, Hangzhou, China) according to the manufacturer’s instructions. Briefly, ATI cells were seeded in each 96-well plate, treated with indicated factors, and further incubated for 24 h or 48 h. Fresh medium containing 10% CCK-8 reagent was added to each well and incubated for 2 h at 37°C. OD450nm value in each well was determined by a microplate reader.
Immunofluorescence staining
Paraffin-embedded lung sections and cultured cells were used for staining. Samples were permeabilized with 0.5% Triton X-100 in PBS for 20 min and blocked with 10% serum (same host as secondary antibody) in PBS 1 h at room temperature. Cells were stained with antibodies against T1a (SC-1666906; Santa Cruz; 1:200), AQP5 (SC-9890; Santa Cruz; 1:200), Cav-1 ( ) as well as proSPC (AB3786; Millipore; 1:1000), KLF2 (BS-2772R; Bioss; 1:200), vimentin (AB8978; Abcam; 1:200), Oct 4 (11263-1-AP; Proteintech; 1:100) and PCNA (AB29; Abcam; 1:1000). Samples were incubated with primary antibody at 4°C overnight followed by the appropriate donkey antimouse, donkey anti-rabbit, or donkey anti-goat secondary antibodies (Invitrogen) protected from light, at room temperature for 1 h. Negative control slides were only exposed to secondary antibodies (data not shown). Nuclei were visualized with DAPI (D8417; Sigma; 0.5 ug/ml).
RNA extraction and quantitative PCR analysis
RNA was isolated from lung tissues or cultured cell preparations using TRIzol (Invitrogen, CA, USA) according to the manufacturer’s instructions. Reverse transcription reactions were performed with Prime Script RT reagent Kit with gDNA Eraser (Perfect Real Time) (Takara, Dalian, China). Gene expressions were determined by quantitative real-time PCR using
SYBR Green Real-Time PCR Master Mix (TOYOBO, OSAKA, Japan). Rat β-actin was used as as internal control. Primer sequences were as follows:
Rat β-actin was used as as internal control. Primer sequences were as follows: Rat β-actin was used as as internal control. Primer sequences were as follows: rat T1a sense: 5'-C A G T G T T G C T C T G G G T T T T G G-3', antisense: 5'- -3'; rat AQP5 sense: 5'-G C T T T G G A A T C A G G C A G A A T G-3', antisense: 5'-A G A C C T G G G T T C A C C A T G T C A-3'; rat proSPC sense: 5'- -3', antisense: 5'-A G C A A G C T G T C G T C C C T T T C-3'; rat Nkx2.1 sense: 5'-A C G T G A G C A A G A A C A T G G C C-3', antisense: 5'-G G T G G T T C T G G A A C C A G A T C-3'; rat KLF2 sense: 5'-C T C A G C G A G C C T A T C T T G C C-3', antisense: 5'-C A C G C T G T T T A G G T C C T C A T C C-3'; rat Snx5 sense: 5'-G C C C G G T T A A A A A G C A A A G A T G T-3', antisense: 5'-G C A T G C T T T A T T T C C A G T T C A G A C A-3'; rat β-actin sense: 5'-A G A G A A G C T G T G C T A T G T T G C C C T-3', antisense: 5'-ACCGCTCATTGCCGATAGTGATGA-3'. Data were processed using 2-ΔΔCT method.
Protein extraction and Western blot analysis
Cultured or transfected cells were lysed in RIPA buffer containing 1% PMSF (P0013, Beyotime). Protein concentration was determined by the BCA protein assay kit (P0012, Beyotime). The protein was loaded onto a SDS-PAGE minigel and transferred onto PVDF membrane. After probed with diluted rabbit antibody at 4°C overnight, the blots were subsequently incubated with HRPconjugated secondary antibody (1:5000). Signals were visualized using ECL Substrates (Millipore, MA, USA). GAPDH was used as an endogenous protein for normalization.
Statistical analyses
All data are expressed as mean ± standard deviation of at least three independent experiments. Differences in measured variables between treated groups were assessed by using the Student’s t-tests or one-way ANOVA. P values less than 0.05 are regarded as significant.
Results
The milieus of damaged lung promote ATIs proliferation
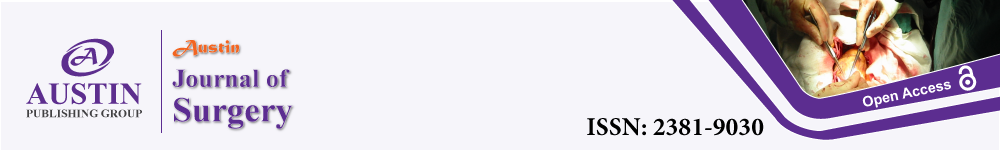
ATIs isolated after 1-2 days were rounded and small with the tendency to group together to form small aggregations. During the following culture, cells spread out, flattened, proliferated, and formed a continuous epithelial monolayer. For the cell identification, cells 4 days after isolation were fixed and immunofluorescence were performed (Figure 1A-1C). By counting nine multiple fields (512um×512um) under microscopy, 89.0±2.1 % of total cells were T1a positive (ATIs specific marker). In accordance with previous studies, our ATIs were proSPC positive in a peri-nuclear distribution, which was very different from the punctate cytoplasmic patterns in ATIIs. To detect the proliferation capability, ATIs were plated at a density of 2 *104/well and observed for 18 days. ATIs became motile at day 4, and proliferated until cells reached confluence. The growth curve was showed in (Figure 1D).
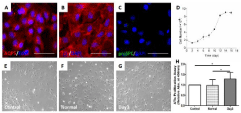
Figure 1: Homogeneity of cultured ATIs and enhanced proliferation capacity
of ATIs after treated with damaged lung milieus.
ATIs exhibited the acceptable homogeneity of morphometric characteristics
in primary culture and expressed the relative lineage markers (A, aquaporin
(AQP)-5; B, T1a; C, proSPC) at 4-culture-day-old. The nuclei were stained
with DAPI. ATIs could continuously proliferate until they were contact
inhibited, and the growth curve was showed here (D) when cells were plated
at 2 *104/well. After treated with lung extracts (100 ug/ml) for 24h, ATIs’
morphologies (E, F and G) and numbers (H) were observed. A significant
difference was observed between control/day3 and normal/day3 groups (*P
< 0.05). Scale bars represent 50 μm.
To mimic ATIs’ in vivo microenvironment, lung extracts were obtained from normal and damaged lung. And their effects on ATIs’ proliferation capacity were compared. As our previous data showed day 3-damaged lung extracts were effectively regulate the characteristics of ATIIs, we explore its potential effect on ATIs. The normal and day 3-damaged lung extracts (100 ug/ml) were added to the ATIs 4-days after isolation, the morphology of ATIs’ were observed and cell numbers were detected via CCK-8 proliferation assay. After 24h, the ATIs treated with lung extracts (Figure 1F,G) elongated slightly comparing to control group (Figure 1E), but there were no sensible differences between normal group and day3 group. The day 3-damaged lung extracts significantly increased the cell numbers by 1.29-fold. The results suggested the day3-damaged lung milieus can promote the proliferation of ATIs.
ATIs exhibit phenotypic changes after treated with damaged lung milieus
We further investigated the phenotypic characteristics in ATIs in response to acute injury lung milieus. There are many types of epithelial cells in alveoli and airway compartments. ATIs and ATIIs are the major epithelium cell types in the alveolar. The latter can express Surfactant Protein C (SPC) and are widely recognized as the progenitor for alveolar epithelium. Clara cells are the main cell type of bronchiolar epithelium and express CCSP (Clara Cell Secretory Protein). They are capable of self-renewal for the maintenance and regeneration of bronchioles based on the classical naphthalene- injury-mode, and regarded as the progenitor cells for bronchiolar epithelium. Based on the characteristics of resistance to naphthalene, a subgroup of clara cells was termed as variant clara cells. Some variant clara cells reside in Bronchioalveolar Duct Junctions (BADJs) and function as stem cells for the repair of bronchiolar epithelium and alveolar epithelium through differentiating into both clara cells and ATIIs. As a member of Nkx factors, which can synergistically regulate tissue-specific gene expression and development in vivo, Nkx2.1 (also known as TTF1 or T/ebp) is previously implicated in lung morphogenesis. As previous articles have demonstrated ATs could express OCT4 in vitro, which belongs to a family of transcription factors containing the POU DNA binding domain and was used as one possible characteristic to identify pulmonary stem cell population. If the ATIs adjust their phenotypes to repair, it is essential to observe how the related genes change.
ATIs maintained their phenotypes under normal culture medium. However, when acute injury lung extracts were added to the medium, the expression of ATIs marker proteins were down-regulated (Figure 2A, D, E), followed by the up-regulation of ATIIs marker proteins (Figure 2B, F) and related transcription factors (Figure 2C). Western blotting further (Figure 2G, H) showed the similar results. According to these results, we suggest that the phenotypic characteristics of ATIs can be regulated by lung extracts. This culture system therefore represents a good model to study how the milieus affects ATIs, particularly how the ATIs react to acute injury lung alveolar milieus.
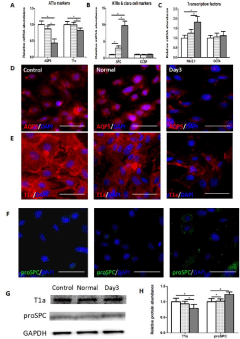
Figure 2: The phenotypic characteristics of ATIs after treated with damaged
lung milieus.
After ATIs were treated with lung extracts (100 ug/ml) for 24h, the expression
of related marker proteins were detected. (A,B and C) Real-time quantitative
PCR analysis (Figure 1A-C) of ATIs marker proteins (AQP5 and T1a), ATIIs
marker protein (SPC), Clara Cell Marker Protein (CCSP) and transcription
factors (Nkx2.1, OCT4). In general, there was a significant decrease of ATIs
markers accompanied by significant increase of ATIIs marker proteins (*P
< 0.05). (D, E and F) Immunofluorescent staining for alveolar epithelial cell
makers in ATIs from three groups. ATIs treated with day3-damaged lung
extracts exhibited lower fluorescence intensity of ATIs marker proteins and
higher fluorescence intensity of ATIIs marker protein comparing with control
group and normal lung extracts. (G, H) Western blot analysis of AQP5, T1a,
and proSPC in treated ATIs. The intensity of proteins bands were analyzed
by Image J software. Scale bars represent 50 μm.
Exposure to damaged lung milieus suppresses KLF2 expression in ATIs
The lung extracts are complex mixtures containing miRNA, cytokines, chemokines and other factors, which could regulated the phenotypes of ATIs through various mechanisms. In previous studies, scientists attempt to analyze the possible effective componenets and mechanisms. Li et al [10] found KLF2 was a critical regulator during the late phase of lung development, which could directly activate the ATIs gene program. Im SK et al. [11] demonstrated that Snx5 was necessary for the differentiation of alveolar epithelial type I cells. Based on these previous data, we tested the expression of KLF2 and Snx5 in ATIs to better understand how lung milieus regulated ATIs’ phenotypes, in other words, how ATIs reacted to lung milieus.
The transcriptional level of KLF2 (Figure 3A) was significantly decreased in day3-damaged milieus group comparing with nontreated (control) or normal lung extracts treated group, whereas the expression of SNX5 (Figure 3B) showed no difference among these groups. Then we explored the protein level of KLF2 in ATIs by immunofluorescent staining (Figure 3C) and western blot (Figure 3D). Consistent with the transcription level, the expression of KLF2 protein showed a similar tendency. So we hypothesized that KLF2 might be one of the regulators associated with the phenotypic module.
Figure 3: Decreased expression of KLF2 in ATIs treated with day3-damaged
lung milieus.
The related genes related with the differentiation of alveolar epithelial type
I cells were detected in ATIs after treated with lung extracts. (A) Real-time
quantitative PCR analysis of KLF2. (B) Real-time quantitative PCR analysis
of SNX5. In comparison with control and normal group, day3 group expressed
lower level of KLF2. (C) Immunofluorescent staining for KLF. (D, E) Western
blot analysis of KLF2 in treated ATIs. Scale bars represent 50 μm.
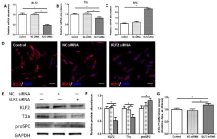
Figure 4: Functional effects of KLF2 downregulation on ATIs.
ATIs were transfected with KLF2 siRNA 24h before the experiments. Control
groups were cultured in normal cultured medium without any treatment, and
NC siRNA groups were transfected with negative control siRNA. (A) Foldchange
of KLF2 in response to gene silencing. (B,C) Expression of T1a and
SPC mRNA following KLF2 knockdown. (D) Immunofluorescent staining for
KLF2 in ATIs after treated with KLF2 siRNAs. (E, F) Western blot analysis of
KLF2, T1a, and proSPC in treated ATIs. (G) Proliferation effects of KLF2 on
ATIs. Scale bars represent 50 μm. Datas are mean±s.e.m. *P < 0.05.
Knockdown of KLF2 expression alters cellular plasticity of ATIs in vitro
To further validate if KLF2 regulates cellular plasticity of cultured ATIs, we tested cell growth and detected cell plasticity when KLF2 gene expression was knocked down by siRNA. Once KLF2 expression was effectively depressed by siRNA (Figure 4A-E), transfected ATIs exhibited increased cell growth (Figure 4G), which was in consistent with the proliferation effects induced by day3-damaged milieus. Furthermore, when ATIs were treated with KLF2 siRNA, we observed synergistic inhibitory effects on T1a expression and promotion effects on proSPC expression, compared to either NC siRNA group or control group. Taken together, these results indicated that KLF2 functions as a potent regulator of the plasticity of cultured ATIs.
Discussion
Increasing progress about cell lineages that contribute toward normal and diseased lung has been made through various recent developments. One of the focus is on comprehensive views of all the differentiated epithelial cells. Dao et al. [12] observed that lung regeneration transcriptomically retrieved lung development at alveoli forming stages after pneumonectomy, which suggested the possible dedifferentiation of differentiated lung cells. Park et al. [13] found the dynamic changes in cell shape and gene expression in ciliated bronchiolar epithelial cells after naphthalene injury, which indicated ciliated cells transdifferentiate into other epithelial cell types. Yadav et al. [14] infect the rats with staphylococcus aureus, they found intermediate cell types expressing both ATI and ATII cell proteins, which indicated the possibility that the cells may originate from ATIs. Jain et al. [15] showed us lineage-labelled Hopx(+) ATIs can proliferate and generate ATIIs following partial pneumonectomy and demonstrate anticipated plasticity of ATIs. These evidences suggest the possibility that ATIs can adjust their plasticity and participate in lung injury repair.
We previously examined the cellular kinetics of ATIs and ATIIs post-acute lung injury systematically and found that alveolar epithelial cells tended to decline at early stage and gradually recovered to a normal level. And when ATIIs were treated with lung extract, they changed their proliferation capacity. All these indicated alveolar epithelial cells were dictated fate by alveolar milieus. We speculate whether the plasticity of ATIs adopt a difference accompanied with damaged lung milieus. So we observed the phenotypes and characteristics of ATIs after treated with lung extract. To our surprise, ATIs proliferated markedly and exhibited features of ATIIs when treated with Day3-milieu, which means the conversion of differentiated state to dedifferentiated state. These results showed the plasticity of ATIs in vitro, which is consistent with recent studies that, although at a low frequency, ATIs could convert to ATIIs and proliferate after pneumonectomy or oncogenic Kras expression. Moreover, we attempted to identify the associated genes that may control the reshaping properties of ATIs or act as signal molecules.
Krüppel-Like Factor 2 (KLF2), also known as Lung Krüppel-Like Factor (LKLF), is a member of the Krüppel-like factor family of zinc finger transcription factors, and it has been demonstrated as a key regulatory molecule during lung development. Pei et al found KLF2 could directly activate the ATIs gene program, and identify its critical role in the maturation of ATIs. Their study also showed KLF2 could directly bind and activate ATIs marker gene promoters, while ATIIs transcription factor Nkx2.1 had no effect. All the results suggested that KLF2 is sufficient to activate ATIs gene program in vitro. Sorting nexin-5 (Snx5) belongs to a large family of proteins that contain the phosphoinositide-binding Phox homology domain and functions in endosomal trafficking of Epidermal Growth Factor Receptor (EGFR) and Cation-Independent Mannose 6-Phosphate Receptor (CI-MPR)/ Insulin-Like Growth Factor II Receptor (IGF2R). Im SK et al. reported that disruption of the Snx5 gene in mice (Snx5-/- mice) resulted in thickened alveolar walls associated with undifferentiated alveolar epithelial type I cells and demonstrated that Snx5 was necessary for the differentiation of alveolar epithelial type I cells. Based on these previous data, we investigated the expression of KLF2 and Snx5 in ATIs treated with damaged lung extract. We found the gene expression of KLF2 was decreased significantly in Day-3 group, while Snx5 was similar in all group. To further detect the relationship between KLF2 and ATIs phenotypes, we knocked down the expression of KLF2 in ATIs and observed the key proteins expression. We found that once KLF2 expression was effectively depressed by siRNA, transfected ATIs exhibited decreased expression of ATIs marker proteins, which was in consistent with those observed in milieu treated cells.
Conclusion
We found significant changes of proliferation and unanticipated plasticity of ATIs treated with damaged alveolar milieus, and also showed that KLF2 has a non-redundant role in regulating ATIs plasticity. It is worth to expect whether ATIs in vivo display similar characteristics to participate in the processes of lung development or repair after injury. As ATIs cover 95-99% of the alveolar surface area, this would provide a substantial cell reservoir with the potential capacity to participate in alveolar regeneration.
References
- Yuan T, Volckaert T, Chanda D, Thannickal VJ, Langhe SP. Fgf10 Signaling in Lung Development, Homeostasis, Disease, and Repair After Injury. Front Genet. 2018; 9: 418.
- Guo L, Karoubi G, Duchesneau P, Aoki FG, Shutova MV, Rogers I, et al. Interrupted reprogramming of alveolar type II cells induces progenitor-like cells that ameliorate pulmonary fibrosis. NPJ Regen Med. 2018; 3: 14.
- Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, et al. Integrin a6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest. 2011; 121: 2855-2862.
- Yang J, Hernandez BJ, Martinez Alanis D, Narvaez del Pilar O, Vila-Ellis L, Akiyama H, et al. The development and plasticity of alveolar type I cells. Development. 2016; 143: 54-65.
- Chen Z, Hu Y, Xiong T, Chen C, Su XX, Huang Y, Zhang L. IL-10 promotes development of acute respiratory distress syndrome via inhibiting differentiation of bone marrow stem cells to alveolar type 2 epithelial cells. Eur Rev Med Pharmacol Sci. 2018; 22: 6085-6092.
- Kim KK, Dotson MR, Agarwal M, Yang J, Bradley PB, Subbotina N, et al. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis. 2018; 9: 1056.
- Li Y, Xu W, Yan J, Xia Y, Gu C, Ma Y, et al. Differentiation of human amniotic fluid-derived mesenchymal stem cells into type II alveolarepithelial cells in vitro. Int J Mol Med. 2014; 33: 1507-1513.
- Archer F, Abi-Rizk A, Desloire S, Dolmazon C, Gineys B, Guiguen F, et al. Lung progenitors from lambs can differentiate into specialized alveolar or bronchiolar epithelial cells. BMC Vet Res. 2013; 9: 224.
- Liu Z, Lai CH, Zhang X, Luo J, Huang X, Qi X, et al. Simvastatin ameliorates total liver ischemia/reperfusion injury via KLF2-mediated mechanism in rats. Clin Res Hepatol Gastroenterol. 2018.
- Li W, Sun M, Zang C, Ma P, He J, Zhang M, et al. Upregulated long noncoding RNA AGAP2-AS1 represses LATS2 and KLF2 expression through interacting with EZH2 and LSD1 in non-small-cell lung cancer cells. Cell Death Dis. 2016; 7: e2225.
- Im SK, Jeong H, Jeong HW, Kim KT, Hwang D, Ikegami M, et al. Disruption of sorting nexin 5 causes respiratory failure associated with undifferentiated alveolar epithelial type I cells in mice. PLoS One. 2013; 8: e58511.
- Dao DT, Anez-Bustillos L, Ourieff J, Pan A, Mitchell PD, Kishikawa H, et al. Heparin impairs angiogenic signaling and compensatory lung growth after left pneumonectomy. Angiogenesis. 2018; 21: 837-848.
- Park KS, Wells JM, Zorn AM, Wert SE, Laubach VE, Fernandez LG, et al. Transdifferentiation of ciliated cells during repair of the respiratory epithelium. Am J Respir Cell Mol Biol. 2006; 34: 151-157.
- Yadav MK, Chae SW, Go YY, Im GJ, Song JJ. In vitro Multi-Species Biofilms of Methicillin-Resistant Staphylococcus aureus and Pseudomonas aeruginosa and Their Host Interaction during In vivo Colonization of an Otitis Media Rat Model. Front Cell Infect Microbiol. 2017; 7: 125.
- Hoffman AM, Shifren A, Mazan MR, Gruntman AM, Lascola KM, Nolen- Walston RD, et al. Matrix modulation of compensatory lung regrowth and progenitor cell proliferation in mice. Am J Physiol Lung Cell Mol Physiol. 2010; 298: L158-L168.