
Case Presentation
Thromb Haemost Res. 2017; 1(1): 1003.
Acquired Haemophilia A: A Serious and Often Unrecognized Disease
Doris Barcellona*, Federica Battolu, Antonella Mameli and Francesco Marongiu
Department of Medical Science and Public Health, University of Cagliari, Italy
*Corresponding author: Barcellona Doris, Department of Medical Science and Public Health, University of Cagliari, 09042 Monserrato, Cagliari, Italy
Received: June 19, 2017; Accepted: August 08, 2017; Published: August 18, 2017
Abstract
Acquired Hemophilia A (AHA) is caused by autoantibodies against factor VIII inducing an increase in the risk of spontaneous bleeding or haemorragies secondary to trauma, surgery or invasive procedure.In about half of the patients the cause of AHA is unknown and the mortality is up to 20%. It is a rare coagulation disorder that affect about 1.5 patients/million/year most of whom are elderly, aged 60 years or more. Haemorrhagic manifestations, located in soft tissues and muscles, expecially in the elderly with a negative bleeding history, concomitantly with a prologed aPTT are the main characteristic of the disesease. In this paper we describe the history of a patient whose diagnosis of AHA has been made with a delay of about five months after several hospital admissions. AHA is not easy to recognize if clinicians did not known the patophysiology, laboratory and clinical appearance of thecoagulative disorders, and diagnosis is often delayed. Early suspicion, investigation, and confirmation of AHA are very important in order to avoid more serious bleeding,invasive approaches and waste of financial resources. Early diagnosis is crucial, and early treatment to control bleeding and to eradicate inhibitors can be life-saving. To help clinicians to prompt identify AHA it should be organized a periodical reminds on this rare disease which can cause mortality if the diagnosis is not made as early as possible.
Keywords: Acquired haemophilia A; Prolonged aPTT; Inhibitor of coagulativefactor VIII
Case Presentation
The patient, a male 84 years old, in January 2017 was hospitalized at the Gastroenterology Unit of a city hospital for the appearance of rectorrhagia and aright extensive subconjunctival hemorrhage. The clinical history showed that the patient was suffering from diverticulosis of the colon and a prostatic adenocarcinoma treated with radiation therapy that had caused an actinic colitis. The followup (8 years) was negative for neoplastic disease. At the admission, the patient presented anemia (Hb 8.5 g/dl) a normal platelet count (259 x 109/L), white blood cells (5.6 x 109/L), and PT INR (1.11) while aPTT was significantly prolonged, 84 seconds with a ratio of 2.9(normal reference values: 20-38 sec and 0.8-1,30 ratio). Colonoscopy showed abundant coagulated blood in the rectum, the sigma and the descending colon with the presence of known diverticula, but the source of bleeding was not identified. The echography of the right ocular bulb showed a bilateral haemorrhagic effusion of about 1.0 cm, without evidence of retrobulbar expansive lesions. The CT total bodyconfirmed the hemorrhagic collections at the right subconjunctival site and showed the presence of mild ascites without evidence of phlogistic lesions, or neoplastic diseases. The patient was transfused with 9 units of packed red cells since anemia worsened and after 22 days of hospitalisation was discharged with the diagnosis of anemia in patients with actinic colitis and diverticulosis of the colon.
After 4 days from the discharge the patient was again hospitalized for intense asthenia and disorientation. Anemia was still present (Hb=8.6 gr/dl), platelet, white blood cells and PT INR were normal but aPTT was further prolonged (127 seconds with a ratio of 4.4). The patient was transfused again with 1 units of packed red cells and again discharged. After 13 days Hb values were unchanged, aPTT was still prolonged (73seconds, ratio 2.5).
In March 2017 the patient came to a Hospital Emergency Room of a city hospital for a head trauma with scalp bruising as a result of an accidental fall and was admitted to the surgery department. The CT of the skull was negative.Four days after, for the appearance of headache, nausea and vomiting, he was submitted to a new CT of the skull that shows asubarachnoidhemorrhage. Hb was low (7.5 gr/dl) but coagulative test were not carried out. The patient was again transfused with 3 packed red cells and transferred to the department of Neurosurgery where the treatment was conservative. CTdocumented an almost total disappearance of subarachnoid bleeding and the persistence of vast bruising of the soft tissues in the left parietal side. Hewas discharged after 9 days with the diagnosis of anemia an cerebral left parietal contusion.
On 29 April 2017 the patient was admitted to the Emergency Department of another city hospital for the appearance of a large hematoma of the left hemithorax and then transferred to the Department of Internal Medicine. At the entrance he showed Hb=6.4 gr/dl, platelet count=176 x 109/L, white blood cells=8.9 x109/L, PT=1.19 INR and aPTT=54 seconds with ratio of 1.9. The patient was tranfused with 6 unit of packed red cells. Hemoglobin raised up to 9.6 gr/dl. Factor VIII was 10 %. The CT total body showed extensive bruising of soft tissues mainly shoulder girdle and in the left hemithorax and a bilateral pleural effusion more evident in the left. Acquired haemophilia was eventually suspected and treatment with methil prednisolone (120 mg/day) was started concomitantly with tranexamic acid, both i.v..
The patient was then transferred to our Internal Medicine Unit. At the entrance the patient showed a extended haematoma (12 cm) of the left breastplate muscle (Figure 1) and extensive bruising of the upper ipsilateral limb and of the left hemithorax, hemorrhagic bubbles were present at both the upper limbs (Figure 2), a vast hematoma was present in the left flank of the abdomen (Figure 3). The bloodchemistry examinations showed Hb=10 gr/dl, platelet count=223 x109/L, PT=1.14 INR and a prolonged aPTT=49 seconds with ratio of 1.62. The mixing test of the aPTT with normal plasma showed no correction of the test that remained prolongedat 120 minutes with a ratio of 1.52. The dosage of factor VIII was 21% while the inhibitor was high (26 Bethesda Units). The therapy with methil prednisolone (120 mg/day) and tranexamic acid (3 gr/day) i.v. was confirmed. After 8 days the patient presented a new episode of rectorrhagia with subsequent anemia, (Hb=8.9 gr/dl), aPTT=55 seconds with ratio of 1.83, and factor VIII of 14%.A new thorax and abdomen CT showed an increase in the size of the large haematoma of the left hemithorax (14x9,5 cm), a bilateral pleural effusion and perihepatic, perisplenic and pelvic effusion. The colonscopy showed multiple telangiectasias of the rectum. He was transfused again with 2 unit of packed red cells. An haemostatic by-passing agent was started (aPCC, FEIBA, 3000 U every 8 hours) associated with Ciclofosfamide 100 mg/day p.o. which was suspended after 13 days for the appearance of bone marrow cytotoxicity. The factor VIII gradually increased and the patient did not show any other bleeding. He was discharged by our department after 34 days,the aPTT was 47 seconds with a ratio of 1.55, factor VIII was 51%. The suggested therapy was metil prednisolone 50 mg/day p.o.and anoutpatient clinic follow-up was planned.
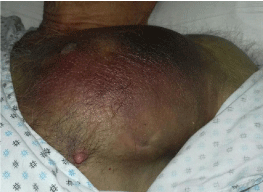
Figure 1: Haematoma of the left breastplate muscle.
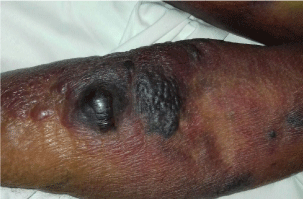
Figure 2: Hemorrhagic bubbles at the upper limb.
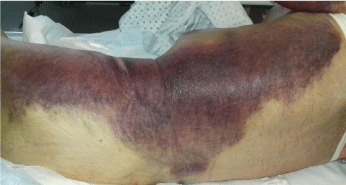
Figure 3: Vast hematoma in the left hemithorax and the left flank of the
abdomen.
Discussion
AHA is induced by inhibitory immunoglobulins (IgG1 and IgG4) against factor VIII. As a conseguence factor VIII is unable to act as a cofactor in the activation of factor X by factor IXa and phospholipids [1]. These antibodies cause a second order non linear inactivation of factor VIII whose residual activity, in general, does not show a relationship with haemorrhagic manifestations nor with the antibodies titer detected with the Bethesda method [2].
AHA increases the risk of spontaneous bleeding or haemorragies induced by trauma, surgery or invasive procedure [3,4]. It is a rare coagulation disorder that affect about 1.5 patients/million/year most of whom are elderly, aged 60 years or more [5]. AHA could be a consequence of underlying pathological conditions such as solid tumors or oncohematological disorders and autoimmune disease but it could also affect women during pregnancy and postpartum period. In about half of the patients the cause of AHA is unknown and the mortality is up to 20% [5]. This case report demonstrates how long may be the delay in the diagnosis of acquired haemophilia. Our patient showed the first haemorragic features in January 2017 but the final diagnosis was made in May 2017, almost five months later. In the last 10 years we have admitted to our department 17 patients with AHA and the main delay of the diagnosis was about 40 days. Since AHA is a rare disease, diagnosis could be difficult for clinicians which are not confident with coagulation disorder and patients must be addressed to a specialized department to have their right diagnosis and appropriate therapy. The main diagnostic problem derives from the ignorance of the existence of the disease and, overall, the lack of any knowledge of the patophysiology, laboratory and clinical appearance of the coagulative disorders. Haemorrhagic manifestations, located in soft tissues and muscles expecially in the elderly with a negative bleeding history concomitantly with a prologed aPTT are the main characteristic of the disesease. aPTT is a routine coagulative test which deserves to be carefully interpreted. It is sensitive to low levels of the intrinsic blood coagulative factors such as XII, XI, IX and VIII. The attention should be focussed on these four coagulative factorsif a normal PT is present. An abnormal aPTT without a recent positive history for haemorrhagic manifestations suggests a defect of either XII or XI factors. The presence of a lupus anticoagulant should also be considered. The latter should be suspected if a mixing text with normal plasma does not correct the basal aPTT values. If a haemorrhagic history is presentand the mixing text with normal plasma is positive, i.e. no correction of the basal aPTT is recorded after 120 minutes, acquired haemophilia is strongly suspected. However, the dosage of factor VIII will confirm this suspicion followed by the determination of the inhibitor with the Bethesda method. The main cause of this drawback in the recognition of this disease lays in the fact that blood coagulation is only a minimal part of university teaching and physicians easily forget the minimum they have learned on this topic. It should be organized periodical reminds on rare disease which can cause mortality if the diagnosis is not quickly made. This concept may be relevant for AHA but we believe it may be important even for other rare disaese which require an early diagnosis. Our patient is an example of a not recognized AHA that fortunately did not lead him to death but was cause of more serious bleeding, repeated hospital admission and important costs for the public healthcare system. If AHA is recognised treatment resides into the eradication of the inhibitor by means of corticosteroids and ciclophosphamide and the employment of haemostatic by-passing agents such as recombinant factor VII (Novoseven, Novo Nordisk) or activated prothrombin complex (FEIBA, Baxalta) [6]. Both these agent are effective in controlling active bleeding. They should be withdrawn when active bleeding disappears because they can cause thrombosis expecially in the elderly population [7]. In 2015 EMA approved the clinical use of Susoctocog alfa(Obizur, Baxalta) a recombinant porcine factor VIII, in which domain B has been substitued with a sequency linker of 24 aminoacids. This chemical procedure confers no reactivity of this new agent with autoantibodies thus controlling bleeding. Kruse- Jarres e coll recently demonstrate the efficacy of Susostocog alfa in the management of 28 patients with AHA [8]. Treatment with by-passing agent should be started as soon as possible if an active bleeding is present. However, any invasive procedure is to be avoided unless absolutely necessary since uncontrollable bleeding may occur [9]. Bypassing agents should be used at a dosage which have been observed to be effective in controlling bleeding: 90 μg/kg of rFVIIa every 2–4 h or 50–100 IU/kg of aPCCs every 8–12 h [6]. However, lower doses of these pro-coagulant agent have been successfully used [10,11].
Conclusion
AHA is a rare disease which requires a rapid recognition and treatment since the mortality rate is high, up to 20 %. Delay in the diagnosis exposes patients to an unacceptable risk of severe bleeding and death. Efforts should be focussed on a better teaching approach to this disease and to blood coagulation pathophysiology and routine coagulative tests.
References
- Franchini M, Lippi G. Acquired haemophilia A. Adv Clin Chem. 2011; 54: 71-80.
- Franchini M, Mannucci PM. Acquired haemophilia A: a 2013 update.Thromb Haemost. 2013; 110: 1114-1120.
- Toschi V, Baudo F. Diagnosis, laboratory aspects and management of acquired hemophilia A. Intern Emerg Med. 2010; 5: 325–333.
- Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol. 2003; 121: 21–35.
- Collins PW, Hirsch S, Baglin TP, Dolan G, Hanley J, Makris M, et al. Acquired hemophilia A in the United Kingdom: a 2-years national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation.Blood. 2007; 109: 1870-1877.
- Huth-Kühne A, Baudo F, Collins P, Ingerslev J, Kessler CM, Lévesque H, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica. 2009; 94: 566-575.
- Hay CR, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors’ Organisation. Br J Haematol. 2006; 133: 591-605.
- Kruse-Jarres R, St-Louis J,Greist A,Shapiro A,Smith H, Chowdary P, et al. Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Hemophilia. 2015; 21: 162-170.
- Collins P, Budde U, Rand JH, Federici AB, Kessler CM. Epidemiology and general guideline soft he management of acquired haemophilia and von Willebr and syndrome.Haemophilia. 2008; 14: 49-55.
- Ma AD, Kessler CM, Al-Mondhiry HA, Gut RZ, Cooper DL. Use of recombinant activated factor VII for acute bleeding episodes in acquired hemophilia: final analysis from the Hemostasis and Thrombosis Research Society Registry acquired hemophilia study. Blood Coagul Fibrinolysis. 2016; 27: 753-760.
- Zanon E, Milan M, Gamba G, Ambaglio C, Saggiorato G, Spiezia L, et al. Activated prothrombin complex concentrate (FEIBA®) for the treatment and prevention of bleeding in patients with acquired haemophilia: as equential study. Thromb Res. 2015; 136: 1299-1302.