
Research Article
Thromb Haemost Res. 2019; 3(2): 1023..
Genetic Depletion of Thromboxane A2 Synthase and Thromboxane-Prostanoid Receptor Signaling Attenuates Ischemia/Reperfusion Induced Oxidative Stress, Inflammation, Apoptosis, Autophagy and Pyroptosis in the Kidney
Chueh TH1, Lin1, Chen KH2,3 and Chien CT1*
¹School of Life Science, National Taiwan Normal University, Taiwan
²Department of Surgery, Far-Eastern Memorial Hospital, Taiwan
³Department of Electrical Engineering, Yuan Ze University, Taiwan
*Corresponding author: Chiang-Ting Chien, School of Life Science, National Taiwan Normal University, No. 88, Section 4, Tingzhou Road, Taipei 11677, Taiwan
Received: April 20, 2019; Accepted: May 14, 2019; Published: May 21, 2019
Abstract
Aims: Enhancement of Thromboxane A2 Synthase (TXAS) activity, Thromboxane A2 (TXA2) release and Thromboxane-Prostanoid receptor (TP) activation leads to severe vasoconstriction and oxidative injury. We explored whether genetic deletion of TXAS/TXA2/TP signaling may reduce renal Ischemia/Reperfusion (I/R) injury in mice.
Methods: Renal hemodynamics and function were evaluated from TXAS+/+TP+/+, TXAS–/–, TP–/– and TXAS–/–TP–/– mice in response to intravenous U46619 (TXA2 mimetic) and I/R injury. We examined renal TXAS and TP expression, BUN and creatinine, Reactive Oxygen Species (ROS) amount, proinflammatory cytokines and pathophysiologic mechanisms including apoptosis, autophagy and pyroptosis under I/R injury.
Results: Renal I/R enhanced the levels of TXAS, TP, nuclear NF-κB, NADPH oxidase gp91, Bax/Bcl-2/Caspase 3/apoptosis, Beclin-1/LC3 II/autophagy, IL- 1β/pyroptosis, renal TXB2 concentration, ROS amount, plasma BUN, creatinine and TXB2 and depressed renal eNOS expression in TXAS+/+TP+/+ mice. All these enhanced parameters were significantly depressed in TXAS–/–, TP–/– and TXAS– /–TP–/– mice. Intravenous U46619 significantly depressed renal microcirculation and enhanced gp91 and Bax/Bcl-2 in TXAS+/+TP+/+ and TXAS–/–, not TP–/– and TXAS–/–TP–/– mice. I/R significantly depressed renal microcirculation in four groups of mice, however, the time for recovery to baseline renal blood flow level was significantly shortened in TXAS–/–, TP–/– and TXAS–/–TP–/– mice vs. TXAS+/+TP+/+ mice. Blocking TXAS/TP signaling also attenuated I/R enhanced pro-inflammatory cytokines profile.
Conclusion: Blockade of TXAS/TXA2/TP signaling confers renal protection against I/R injury through the action of anti-oxidation, anti-inflammation, antiapoptosis, anti-autophagy and anti-pyroptosis.
Keywords: Apoptosis; Autophagy; Pyroptosis; Thromboxane A2 synthase; Thromboxane-prostanoid receptor
Abbreviations
COX-1: Cyclooxygenase-1; COX-2: Cyclooxygenase-2; eNOS: endothelial Nitric Oxide Synthase; ES: E14TG2a Embryonic Stem; I/R: Ischemia/Reperfusion; KO: Knockout; NO: Nitric Oxide; O2-: Superoxide Anion; ROS: Reactive Oxygen Species; TXAS: Thromboxane A2 Synthase; TXAS–/–: TXAS-Knockout; TXA2: Thromboxane A2; TXB2: Thromboxane B2; TP: Thromboxane- Prostanoid Receptor; TP–/–: TP-Knockout; TXAS–/–TP–/–: TXAS and TP Double-Knockout
Introduction
Thromboxane A2 (TXA2) is a member of the prostanoid family of arachidonic acid metabolites generated by the sequential action of phospholipase A2, Cyclooxygenase-1 (COX-1), Cyclooxygenase-2 (COX-2) or TXA2 Synthase (TXAS). The very unstable TXA2 regulates multiple biological processes via its specific thromboxaneprostanoid (TP) receptor to stimulate platelet aggregation and vasoconstriction in vascular and respiratory smooth muscles [1- 4]. The cellular and tissue distribution of TP receptors is closely correlated with that of TXAS [5]. TXA2 [6] as well as isoprostanes [7,8], nonenzymatic free radical-derived products of arachidonic acid, that can activate the TP in vivo [9], is highly elevated in renal, myocardial infarction, atherosclerosis, stroke, bronchial asthma and preeclamptic placenta [1,10-13]. In addition, TXAS and TP are highly expressed within the atherosclerotic lesion areas [14] and during vascular and atherothrombotic diseases [15].
Renal Ischemia/Reperfusion injury (I/R) frequently occurs after infarction, sepsis and organ transplantation and exacerbates kidney dysfunction initiating an inflammatory cascade including Reactive Oxygen Species (ROS), cytokines/chemokines, and leukocytes activation/infiltration [16-18]. Renal TXA2 is elevated in cyclosporine treated kidneys [19], nephritic glomeruli [10,20] and I/R injury [21] reflecting an enhanced TXAS/TXA2/TP signaling to impair the kidney. TXA2 release activates TP via PKC-ζ-mediated NADPH oxidase enhancement [15] and increases O2·- and ONOO-, resulting in endothelial Nitric Oxide Synthase (eNOS) uncoupling in endothelial cells [22]. Anoxia/reoxygenation evokes overproduction of platelet TXB2 and isoprostanes through the action of NADPH oxidase-dependent ROS generation [23]. Targeting the TP or TXAS effectively improves renal function and pathology in humans [21,24]. In animal models, aspirin decreases the severity of renal I/R injury and the development of tubular atrophy [25]. In clinical trials, preoperative low dose aspirin treatment reduced postoperative acute kidney injury, decreased hemodialysis requirements and decreased postoperative hospital stay without increasing bleeding [26]. Some TXAS inhibitors offer an advantage over aspirin in that they may redirect arachidonate metabolism towards PGI2 and other protective eicosanoids [27]. TXAS inhibitors used together with TP receptor antagonists exert a greater anti-platelet effect than low-dose aspirin therapy [28]. However, despite promising results from animal models, the clinical efficacy of TXAS/TP inhibitors is not conclusive and there are even findings suggesting that these agents may inhibit platelet aggregation via other mechanisms besides inhibiting TXA2 activity [3]. Thus, mice with double deletions of TXAS and TP receptors are valuable not only for investigating the phenotype of complete absence of TXA2 activity but also for exploring the pharmacological mechanisms related to these dual inhibitory agents.
In response to oxidative injury, renal cells displayed three types of programmed cell death including apoptosis, autophagy, and pyroptosis [18,29]. We hypothesize these types of programed cell death may be involved in I/R enhancing TXAS/TXA2/TP signalinginduced renal dysfunction. To explore the TXA2 effects and mechanisms on I/R induced autophagy, apoptosis and pyroptosis formation in the damaged kidney is important for alleviating and preventing TXA2-induced renal dysfunction. This study was the first use of four types of targeting gene depleted mice, including TXAS+/+TP+/+, TXAS–/–TP+/+, TXAS+/+TP–/–, TXAS–/–TP–/– mice to explore the effect of blocking TXAS/TXA2/TP signaling on renal I/R injury.
Materials and Methods
Animals
Thromboxane A synthase knockout (TXAS–/–) mice: The TXAS– /– mice are provided by Dr. Shu-Wha Lin’s group from National Taiwan University College of Medicine. A targeting vector is designed to replace the distal portion exon 9 of the endogenous gene with a phosphoglycerate kinase-hypoxanthine phosphoribosyltransferase) cassette. This construct is electroporated into E14TG2a Embryonic Stem (ES) cells. Correctly targeted ES cells are injected into C57BL/6J blastocysts. Chimeric mice are bred with C57BL/6J for 10 generations, the homozygous TXAS–/– mice were obtained by intercrossing the heterozygous TXAS+/– mice.
Thromboxane A2 receptor knockout (TP–/–) mice: The TP–/– mice are generated by Dr. Thomas M. Coffman’s group and also provided by Dr. Su-Wha Lin. A neomycin resistance gene driven by the phosphoglycerate kinase promoter is inserted into a unique SfiI site near the proximal end of exon 2. This insertion targeting vector disrupts the coding sequence of the TP gene in the third transmembrane domain. This construct is electroporated into ES cells. Correctly targeted ES cells are injected into C57BL/6J blastocysts. Chimeric mice are bred with C57BL/6J for 10 generations, the homozygous TP–/– mice are obtained by intercrossing the heterozygous TP+/– mice.
Thromboxane A2 synthase and thromboxane A2 receptor double knockout (TXAS–/–TP–/–) mice: TXAS–/– and TP–/– mouse in the C57BL/6 background are crossbred to generate TXAS–/–TP–/– double knockout mice. Male and female mice with or without targeting gene depletion, including TXAS+/+TP+/+, TXAS–/–TP+/+, TXAS+/+TP–/– and TXAS–/–TP–/– mice (B6 background), have been developed and are provided from National Taiwan University core laboratory. The mice were housed at the Experimental Animal Center of National Taiwan University College of Medicine, with a temperature- and humidityregulated environment (22±2°C, 55±5% RH) and a consistent light cycle (light from 07:00 to 18:00 o’clock). Standard powdered diet containing 58% of carbohydrates, 28.5% of proteins and 13.5% of fat (Laboratory Rodent diet 5001, Young Li Trading Company, LTD, Sijhih City, New Taipei City, Taiwan) and tap water were provided ad libitum. All surgical and experimental procedures are approved by Institutional Animal Care and Use Committee of National Taiwan Normal University and are in accordance with the guidelines of the National Science Council of Republic of China (NSC 1997).
Renal ischemia/reperfusion (I/R)
TXAS+/+TP+/+, TXAS–/–TP+/+, TXAS+/+TP–/– and TXAS–/–TP–/– mice with or without renal I/R (n = 6 in each group) were anesthetized with intraperitoneal urethane (1.2 g/kg). The trachea was exposed via a midline cervical incision and intubated. PE-10 catheters were placed in the left carotid artery for measurement of the heart rate and arterial blood pressure by an ADI system (Power-Lab/16S) with a transducer (P23 1D, Gould-Statham, Quincy, USA), and in the jugular vein for administration of test agents. Renal I/R was induced in the mice as described below. For induction of ischemia, the bilateral renal arteries were clamped 45 min with a small vascular clamp. Sham-operated animals underwent similar operative procedures without occlusion of the renal arteries. Reperfusion was initiated by removal of the clamp for 4 hours. After I/R insults, arterial blood and urine were collected for renal functional determination (Blood Urea Nitrogen (BUN) and creatinine) and plasma and renal TXB2 determination (an enzyme immunoassay, Cayman Chemical, Ann Arbor, MI) [30]. After sacrifice with intravenous KCl, these two kidneys were resected and divided into two parts. One part was stored in 10% neutral buffered formalin for immunohistochemistry and in situ pathologic assay, and another was quickly frozen in liquid nitrogen and stored at –70oC for protein isolation.
Renal microcirculation determination
To examine the in vivo response of renal arterial constriction to U46619 and renal I/R, a full-field laser perfusion imager (MoorFLPI, Moor Instruments Ltd., Devon, UK) was used to continuously quantitate the renal microcirculatory blood flow intensity [18]. In brief, the imager used laser speckle contrast imaging, which would exploit the random speckle pattern generated when tissue was illuminated by laser light. The random speckle pattern changed when blood cells moved within the Region Of Interest (ROI). When there was a high level of movement (fast flow), the changing pattern became more blurred, and the contrast in that region reduced accordingly. The contrast image was processed to obtain a 16-color coded image that correlated with blood flow in the heart. Blue was defined as low flow and red as high flow. The microcirculatory blood flow intensity of each ROI was recorded as Flux with perfusion unit, which was related to the product of average speed and concentration of moving red blood cells in the kidney sample volume. The negative control value was set at 0 perfusion unit (blue color) and the positive value was at 1000 perfusion unit (red color). The perfusion units were realtime analyzed by the MoorFLPI software version 3.0.
In vivo renal ROS detection
We directly measured the renal ROS in response to renal I/R in vivo via an intravenous infusion of a superoxide anion probe, 2-Methyl-6-(4-methoxyphenyl)-3,7-dihydroimidazo-[1,2-a]- pyrazin- 3-one-hydrochloride (MCLA) (0.2 mg/mL/hour, TCIAce, Tokyo Kasei Kogyo Co. Ltd., Tokyo, Japan) and detected by a Chemiluminescence Analyzing System (CLD-110, Tohoku Electronic In. Co., Sendai, Japan) [16]. In brief, after surgery, the rat was maintained on a respirator and a circulating water pad at 37°C during photon detection. For excluding photon emission from sources other than the kidney, the anesthetized animal was housed in a dark box with a shielded plate. Only the kidney was left unshielded and was positioned under a reflector, which reflected the photons from the exposed kidney surface onto the detector area. The MCLA-enhanced chemiluminescent signal from the kidney surface was recorded continuously by the chemiluminescence analyzer. The total ROS value was measured by area under curve from the kidney. The chemiluminescent signal obtained from 0.2 mL saline in 1 mL of MCLA (0.2 mg/ml) or 0.2 ml xanthine (0.75 mg/kg body weight)/ xanthine oxidase (24.8 mU/kg body weight) in 1 ml of MCLA (0.2 mg/ml) was regarded as negative or positive control.
Western blotting
The kidneys were ground to powder in liquid nitrogen and then the powder was lysed in RIPA Buffer (Bio Basic, NY, USA) supplemented with protease inhibitor (Roche, Basel, Switzerland) for 10 minutes at 4°C. Detection of signals was performed by western lightning plus-ECL (PerkinElmer, Waltham, USA). The expression levels of TXAS, TP, NF-κB, gp91, apoptosis-related proteins including Bcl-2, Bax, Caspase 3, autophagy-related proteins Beclin-1 and LC3-II and pyroptosis-related protein IL-1β were analyzed by Western blotting in kidney tissues. The Western blotting method has been described elsewhere [18].
Antibodies raised against TXAS (Cayman), TP (Cayman), NF-κB (R&D Systems), NADPH oxidase (gp91phox; Santa Cruz Biotechnology), Beclin-1 (Cell Signaling Technology, Inc., Danvers, MA), LC3-II (Cell Signaling Technology, Inc.), Bax (Chemicon), Bcl- 2 (Transduction, Bluegrass-Lexington, KY), caspase-3 (Cell Signaling Technology, Denver, USA) and β-actin (Sigma, Saint Louis, MI) were used. The density of the band with the appropriate molecular mass was determined semi-quantitatively by densitometry using an image analyzing system (Alpha Innotech, San Leandro, CA).
Secondary antibodies included HRP-conjugated goat anti-mouse IgG, HRP-conjugated rabbit anti-goat IgG, and HRP-conjugated goat anti-rabbit IgG (all for 1:10000; all from SouthernBiotech Laboratories, Birmingham, USA). For quantitative comparison of the protein expression levels, intensities of specific bands, corresponding to the proteins of interest were measured using densitometry analysis.
Histologic studies
Tissue samples were fixed in 4% paraformaldehyde and embedded in paraffin. We obtained 5-μm sections and stained them with the hematoxylin and eosin. We calculated the mean renal injury score in each mouse with light microscope and averaged the scores for each group. Twenty tubules in each kidney were randomly selected at a × 200 magnification, and the degree of renal damage was scored using the scoring system for renal injury reported by Wagner et al. (2003) [31]. The percentage of tubular injury parameters containing epithelial flattening, tubular atrophy, tubular dilatation, and brush border loss were estimated by a pathologist who was blind to the identity of the specimen using a 4-point scale in ten randomly chosen, nonoverlapping fields. Degree of injury was graded on a scale from 0 to 4: 0 = normal; 1 = mild, involvement of less than 25% of the cortex; 2 = moderate, involvement of 25 to 50% of the cortex; 3 = severe, involvement of 50 to 75% of the cortex; and 4 = extensive damage involving > 75% of the cortex.
Detection of autophagy, apoptosis and pyroptosis in the I/R kidneys
To examine the effect of TXAS or TP knockout on several oxidative stress parameters, we performed LC3-II-related autophagy, Caspase 3/terminal deoxynucleotidyl Transferase-Mediated Nick- End Labeling (TUNEL) apoptosis method [18] and IL-1β-mediated pyroptosis to investigate the presence and extent of three types of programmed cell death in renal I/R injury. The renal sections (5 μm) were prepared, deparaffinized, and stained by the hematoxylin & eosin, LC3-II stain, TUNEL-avidin-biotin-complex methods and IL- 1β stains. A biotinylated secondary antibody (Dako, Botany, NSW, Australia) was then applied followed by streptavidin conjugated to HRP (Dako). The chromogen used was Dako Liquid diaminobenzene (DAB). Twenty high-power (×400) fields were randomly selected for each section, and the value of each oxidative stress was analyzed using a Sonix Image Setup (Sonix Technology Co., Ltd) containing image analyzing software Carl Zeiss AxioVision Rel.4.8.2 (Future Optics & Tech. Co. Ltd., Hangzhou, China). The apoptotic index was calculated as the number of TUNEL-positive nuclei per high-power field (×400).
Statistical analysis
All values were expressed as mean ± Standard Error Mean (SEM). Differences within groups were evaluated by paired t-test. One-way analysis of variance was used for establishing differences among groups. Intergroup comparisons were made by Duncan’s multiplerange test. Differences were regarded as significant if P ‹ 0.05 is attained.
Results
Blocking TXAS/TP signaling reduces I/R induced oxidative stress
The original graph of renal I/R on TXAS, TP, NADPH oxidase gp91 ans eNOS expression by western blotting was displayed in Figure 1A. Renal I/R significantly enhanced TXAS, TP and NADPH oxidase gp91 and depressed eNOS expression as compared to the baseline control in the TXAS+/+TP+/+ kidney (Figure 1B). The increased renal TXAS, TP and gp91 expressions responding to I/R were significantly decreased in the TXAS–/–, TP–/– or TXAS–/–TP–/– mice (Figure 1B). The degree of decreased renal eNOS by I/R was significantly preserved in the TXAS–/–, TP–/– or TXAS–/–TP–/– mice as compared to TXAS+/+TP+/+ mice. We further compared the plasma and kidney TXB2 level before and after renal I/R injury. Renal I/R significantly increased plasma (Figure 1C) and kidney TXB2 concentration (Figure 1D) in the TXAS+/+TP+/+ and TXAS+/+TP–/– mice. However, renal I/R did not significantly increase plasma and kidney TXB2 level in the TXAS–/– and TXAS–/–TP–/– mice. We used immunofluorescence stain to confirm the higher fluorescent intensity of TXAS or TP in the TXAS+/+TP+/+ mice with I/R injury but less expression of TXAS or TP fluorescence in TXAS–/–, TP–/– or TXAS–/–TP–/– mice subjected to I/R injury (Figure 1E). These data implicate blocking TXAS/TP signaling may reduce gp91 mediated oxidative stress in the kidney.
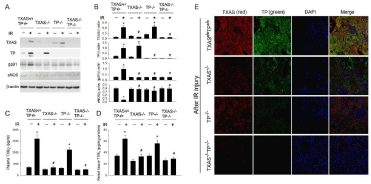
Figure 1: By using Western blotting analysis, renal Ischemia/Reperfusion (IR) markedly increased TXAS, TP and NADPH oxidase gp91 expression and depressed
eNOS expression in the TXAS+/+TP+/+ kidney (A), however, the increased TXAS, TP and gp91 expressions and suppressed eNOS expression were significantly
attenuated in the TXAS-/-, TP-/- or TXAS-/-TP-/- mice (B). The plasma (C) and renal tissue (D) concentration of TXB2 before and after IR was displayed. The use of
immunofluorescence stain consistently evidenced the high expression of TXAS or TP in the TXAS-/-TP-/- mice with IR injury but less expression of TXAS or TP in
TXAS-/-, TP-/- or TXAS-/-TP-/- mice subjected to IR injury (E). *P ‹ 0.05 vs. sham TXAS+/+TP+/+ mice. # P ‹ 0.05 vs. IR TXAS+/+TP+/+ mice.
U46619 reduces renal microcirculation and induces apoptosis and oxidative stress
To study the effect of TXA2 on renal microcirculation, we intravenously infused U46619, a compound known as a TXA2 agonist, into these transgenic mice. As shown in Figure 2, in the absence of U46619, the kidney displayed a red color with high perfusion units in all mice. Fifty-min intravenous U46619 (0.04 mg/ min/kg BW) reduced the renal microcirculation and the perfusion units in B6 mice and partly in TXAS–/– mice. In contrast, there was no significant change in the microcirculation from TP–/– and TXAS–/– TP–/– mice (Figure 2B). We further determined the effect of U46619 infusion on renal Bax, Bcl-2, Caspase 3 and NADPH oxidase gp91 expression (Figure 2C) in the kidneys of four types of mice. The immunofluorescent bax and immunohistochemical Caspase 3 and gp91 were highly expressed in the B6 TXAS–/– kidneys after 4 h of U46619 treatment. The Bax, Caspase 3 and gp91 were less expressed in the U46619 treated kidneys of TP–/– and TXAS–/–TP–/– mice.
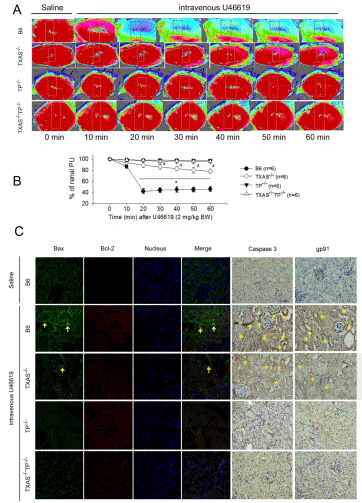
Figure 2: The effect of U46619 infusion on renal microcirculation (A,B), immunofluorescence of Bax and Bcl-2 expression and immunohistochemistry of Caspase
3 and NADPH oxidase gp91 (C) in the kidneys of four types of mice. The immunofluorescent bax and immunohistochemically brown Caspase 3 and gp91 are
indicated with yellow arrows in the kidneys after 4 h of U46619 treatment. *P ‹ 0.05 vs. B6 (TXAS+/+TP+/+) mice. a P ‹ 0.05 vs. TXAS-/- mice.
Deleted TXAS/TP reduced inflammation, apoptosis, autophagy and pyroptosis
We explored the effect of genetic deletion of TXAS/TP on renal I/R evoked inflammatory transcription factor (NF-κB), pyroptosisrelated IL-1β, autophagy-related Beclin-1 and LC3-II, apoptosisrelated Bax, Bcl-2 and caspase 3 proteins expression in four phenotypes of mice. The basal level of these proteins was similar in four types of mice (Figures 3A-3D). In response to the I/R injury, the lower expression of NF-κB, IL-1β, Beclin-1, LC3-II, Bax, and caspase 3 was found in TXAS–/–, TP–/– or TXAS–/–TP–/– mice as compared to B6 mice (Figures 3E-3L). These data implicate blocking TXAS/ TP signaling attenuated I/R induced inflammation, pyroptosis, autophagy and apoptosis in the damaged kidneys.
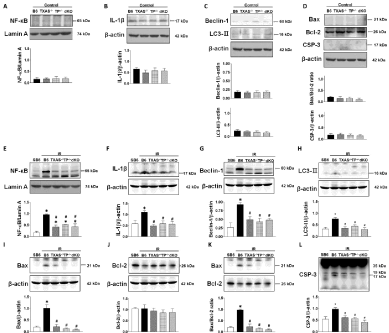
Figure 3: Effect of TXAS or TP knockout on renal Ischemia/Reperfusion (IR) evoked inflammatory transcription factor (NF-κB), pyroptosis-related IL-1β, autophagyrelated
Beclin-1 and LC3-II, apoptosis-related Bax, Bcl-2 and caspase 3 (CSP-3) proteins expression in four phenotypes of mice. The basal level of these proteins
is indicated in A-D. In response to the IR injury, these original graphs and statistic data of these proteins are displayed in E-L. *P ‹ 0.05 vs. sham B6 (SB6) mice.
# P ‹ 0.05 vs. IR B6 (TXAS+/+TP+/+) mice.
Figure 4A demonstrated that in response to renal I/R, high expression of Bax, very low expression of Bcl-2 and increased TUNEL positive cells in the kidney were found in B6 (TXAS+/+TP+/+) mice. As compared to TXAS+/+TP+/+ mice, renal Bax expression was lower expressed in the TXAS–/– (Figure 4B), TP–/– (Figure 4C) or TXAS–/– TP–/– mice (Figure 4D). Higher renal Bcl-2 expression was found in the TXAS–/–, TP–/– or TXAS–/–TP–/– mice as compared to TXAS+/+TP+/+ mice. I/R significantly increased TUNEL positive cells in four groups of mice, however, the increased TUNEL cells (Figure 4E), BUN (Figure 4F) and creatinine level (Figure 4G) by I/R was significantly decreased in TXAS–/–, TP–/– or TXAS–/–TP–/– mice.
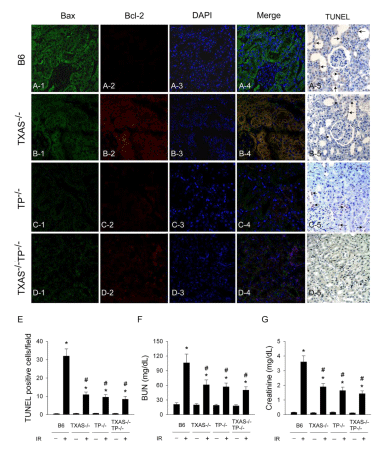
Figure 4: In response to renal Ischemia/Reperfusion (IR), high expression of Bax, very low expression of Bcl-2 and increased TUNEL positive cells in the kidney
were found in B6 (TXAS+/+TP+/+) mice. When compared to TXAS+/+TP+/+ mice, renal Bax expression was lower expressed in the TXAS-/- (B), TP-/- (C) or TXAS-/-TP-/-
mice (D). Higher renal Bcl-2 expression was found in the TXAS-/-, TP-/- or TXAS-/-TP-/- mice. IR significantly increased TUNEL positive cells in four groups of mice,
however, the increased TUNEL cells (E), BUN (F) and creatinine level (G) by IR was significantly depressed in TXAS-/-, TP-/- or TXAS-/-TP-/- mice. *P ‹ 0.05 vs.
respective sham mice. # P ‹ 0.05 vs. IR B6 (TXAS+/+TP+/+) mice.
As shown in Figure 5, in response to renal I/R, higher expression of tubular injury score, IL-1β, LC3 II and Caspase 3 were found in B6 (TXAS+/+TP+/+) kidneys as compared to their controls. These oxidative parameters were significantly attenuated in the TXAS–/–, TP–/– or TXAS–/–TP–/– kidneys vs. TXAS+/+TP+/+ kidneys.
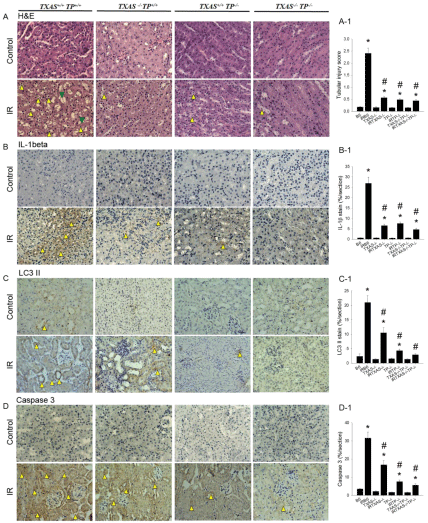
Figure 5: In response to renal Ischemia/Reperfusion (IR), high expression of tubular injury score (A, A-1), IL-1β (B, B-1), LC3 II (C, C-1) and Caspase 3 (D, D-1)
in the kidney were found in B6 (TXAS+/+TP+/+) mice as compared to their controls. These oxidative parameters were significantly attenuated in the TXAS-/-, TP-/- or
TXAS-/-TP-/- kidneys vs. TXAS+/+TP+/+ kidneys. *P ‹ 0.05 vs. sham B6 mice. # P ‹ 0.05 vs. B6 mice subjected to I/R injury.
In response to renal I/R, the renal microcirculation was determined with a moor image in these four groups of mice (Figure 6A). Renal I/R significantly depressed the renal microcirculation indicated by the decreased perfusion unit in four types of mice (Figure 6B). The depressed renal PU by I/R was significantly recovered toward normal value within 10-min reperfusion in the TXAS–/–, TP–/– or TXAS–/– TP–/– mice vs. B6 mice. The blood flow recovery time (the time for depressed PU return to baseline PU value) was displayed in an order of B6 › TXAS–/– › TP–/– › TXAS–/–TP–/– mice (Figure 6C).
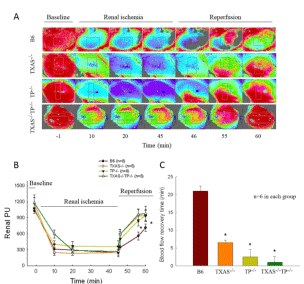
Figure 6: In response to renal Ischemia/Reperfusion injury (I/R), the renal microcirculation was determined with a moor image in these four groups of mice as
demonstrated in A. Renal I/R significantly depressed the renal microcirculation indicated by perfusion unit (PU) in B6 (TXAS+/+TP+/+) mice when compared to TXAS-
/- mice, TP TP-/- mice , or TXAS-/-TP-/- mice (B). The renal PU significantly recovered toward normal value within 10-min reperfusion in the TXAS-/-, TP TP-/- , and
TXAS-/-TP-/- mice. The blood flow recovery time was displayed in C. *P ‹ 0.05 vs. B6 mice.
Blocking TXAS/TP signaling reduced I/R evoked ROS
We determined the renal I/R effect on whole blood ROS in vitro and kidney ROS in vivo among these four groups of mice in the Figure 7. The original data of whole blood ROS from pre- and post-I/R of four types of mice was demonstrated in Figure 7A. Figure 7B showed that, the whole blood ROS level from post I/R was significantly higher (P‹0.05) than that from pre-I/R in the TXAS+/+TP+/+ mice. However, the blood ROS value between pre- and post-I/R was similar in the TXAS–/–, TP–/– or TXAS–/–TP–/– mice. Figure 7C showed the original data of renal ROS in response to I/R in the four types of mice. Figure 7D displayed that in response to I/R, the in vivo kidney ROS level was significantly elevated as compared to baseline level in TXAS+/+TP+/+ mice than that in the TXAS–/–, TP–/– or TXAS–/–TP–/– mice. However, the kidney ROS level was significantly depressed in the TXAS–/–, TP–/– or TXAS–/–TP–/– mice compared to that in TXAS+/+TP+/+ mice.
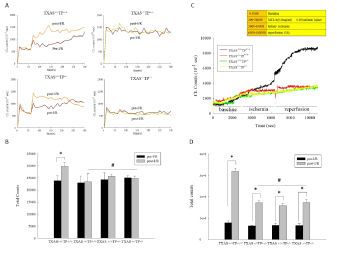
Figure 7: The original data of in vitro blood ROS before (pre-I/R) and after renal ischemia/reperfusion injury (post-I/R) was shown in A. Post I/R, the in vitro whole
blood ROS level was significantly elevated in TXAS+/+TP+/+ mice than that in the TXAS-/-, TP-/- or TXAS-/-TP-/- mice (A, B). The original data in in vivo kidney ROS level
was demonstrated in C. The statistic data showed that the in vivo kidney ROS level was significantly higher after renal I/R in these four groups of mice (D). However,
the kidney ROS level was significantly depressed in the TXAS-/-, TP-/- or TXAS-/-TP-/- mice compared to that in TXAS+/+TP+/+ mice (D). *P ‹ 0.05 vs. respective sham
mice. # P ‹ 0.05 vs. IR TXAS+/+TP+/+ mice.
Effect of blockade of TXAS/TP signaling on renal I/Rinduced inflammatory cytokines
To determine the effect of I/R on proinflammatory cytokines profile in four groups of mice, we determined the plasma before and after renal I/R injury. After I/R injury, nineteen plasma cytokines were up-regulated and 14 cytokines were down-regulated in TXAS+/+TP+/+ mice (Figure 8). We also found that among these 19 up-regulated cytokines (SDF-1 alpha, MCP-1, GCSF, TECK, TARC, MCP-5, TIMP-1, BLC, MIP-1 gamma, TF alpha, MIG, IGFBP 6, sTNFRI, CD40, Axl, CD30, CRG-2, VEGF, and VCAM-1), blockade of TXAS/ TP signaling by knockout TXAS or TP markedly reduced these 19-upregulated cytokines in the TXAS–/–, TP–/– or TXAS–/–TP–/– mice. However, among 14 down-regulated cytokines (IL-4, IL-5, IL-6, Fas Ligand, CX3CL1, IL-17, IL-12 p40/70, IL-10, IL-13, Leptin R, Leptin, IL-3, LIX, L Selectin and IL-3 Rb) in TXAS+/+TP+/+ mice, blocking TXAS/TP in the TXAS–/–, TP–/– or TXAS–/–TP–/– mice displayed a similar pattern with TXAS+/+TP+/+ mice.

Figure 8: Determination of multiple cytokines by cytokine antibody array in the plasma before and after renal I/R injury of the TXAS+/+TP+/+, TXAS-/-, TP-/- or TXAS-/-
TP-/- mice (A). The respective expression of each cytokine dot was indicated in the array (B). The statistic data for 19 up-regulated cytokines (up arrows), 14 downregulated
cytokines (down arrows) and 6-unaffected cytokines in TXAS+/+TP+/+ mice was demonstrated in C. Note that blockade of TXAS/TP signaling potentially
reduced the dot-spot intensity of 19 up-regulated proinflammatory cytokines.
Discussion
TP mRNA is most abundantly expressed in the glomerulus, followed by the distal convoluted tubule › proximal tubule, thick ascending limb of Henle, and outer medullary collecting duct [32]. TP proteins are expressed both in glomeruli and in tubules prominently at the base of the brush border of proximal tubules and at the luminal surface of thick ascending limbs and distal convoluted tubules and its overexpression contributes to nephritic glomeruli [33]. Renal I/R primarily attacked the renal proximal and distal tubules through the action of excess reactive oxygen species production [16]. Our data in Figure 1 directly demonstrated for the first time the blockage effect of TXAS/TXA2/TP signaling on renal I/R induced renal tubular dysfunction in TXAS+/+TP+/+, TXAS–/–, TP–/– and TXAS–/–TP–/– mice generated by gene targeting. Our data showed that bilateral renal I/R significantly decreased renal microcirculation and eNOS expression and elevated TXAS, TP and NADPH oxidase gp91 protein expression in the TXAS+/+TP+/+ mice. Renal I/R increased urinary TXB2 and O2-. levels in TXAS+/+TP+/+ and TP–/–, but not in TXAS–/– and TXAS–/– TP–/– mice during post-reperfusion stage. The decreased level of U46619 and I/R-induced renal microcirculation was all significantly recovered in TXAS–/–, TP–/– and TXAS–/–TP–/– mice compared to TXAS+/+TP+/+ mice. TXAS knockout does not affect basal TXB2 but depresses renal I/R elevated TXB2 concentration in our data. Inhibition of TXAS expression to synthesize TXA2 and TP activation through TXAS or TP depletion confers renal protection against I/R injury. Our data also evidenced that the enhanced TXAS/TXA2/TP signaling was associated with the increased gp91 expression, ROS amount, apoptosis, autophagy and pyroptosis formation in the renal tubules. Blockade of TXAS/TXA2/TP signaling with gene targeting significantly reduces renal I/R injury. A schematic summary is illustrated in Figure 9.
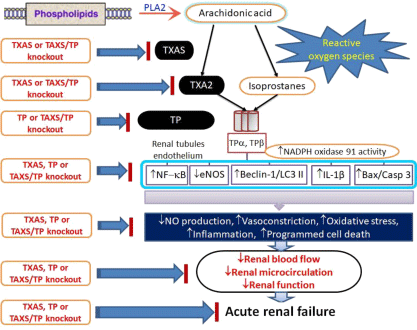
Figure 9: A schematic summary of the Thromboxane A2 (TXA2) signaling pathway involving TXA2, TXA2 synthase (TXAS), and Thromboxane-Prostanoid receptor
(TP). The signaling pathway of renal ischemia/reperfusion injury in genetically depleted TXAS-TP mice.
How did the enhancement of TXAS/TXA2/TP signaling evoke oxidative injury? The TP expression and TXA2 release are elevated during vascular and atherothrombotic diseases associated with the increased ROS release and the reduced cyclic GMP-mediated NO bioactivity [15]. TP activation by TXA2 agonist U46619 induced renal vasoconstriction through the increased CD38 and p67(phox)-mediated O2·- formation and the suppressed eNOS activity in renal resistance arterioles [34]. Pharmacological or genetic inhibition of TP abolished U46619-induced O2·- formation and PKC-ζ inhibition abolished TP activation-induced membrane translocation of p67(phox) and O2·- production [34]. Pharmacologic inhibition of TXA2 release from total saponin (Korean red ginseng) inhibited COX-1 and TXAS activities in platelet microsomes to improve thrombotic diseases [35]. On the other hand, platelet isoprostane, 8-isoprostaglandin F2a (8-iso- PGF2a), a proaggregating molecule, maximally derived from gp91 activation and was inhibited -8% by aspirin and -58% by a specific inhibitor of gp91 [36]. In acute kidney injury, PKC-e-deficiency markedly improved survival rate and attenuated kidney dysfunction through anti-apoptotic and anti-inflammatory signaling [37]. TP agonist U46619 administration caused transient increases in blood pressure followed by cardiovascular collapse and shock in wild-type mice, but did not affect hemodynamics and arachidonic acid-induced shock in TP–/– mice [38]. These genetically depleted TXAS–/–, TP–/– and TXAS–/–TP–/– mice exhibited more stable vasoactive responses in the presence of different vasoconstrictors or vasodilators in the mesenteric arterioles [4]. Our previous results found gene deletion of TP protects from the ET-1-mediated reduction in cardiac perfusion and preserves eNOS expression and NO activity in TP–/– and TXAS–/– TP–/– mice [4]. TXAS deficiency does not confer the same protection against I/R injury as TP deficiency, which may point to an increase in the production of TP agonists like isoprostanes or Prostaglandins (PGF2a) other than TXA2 in the I/R injury.
TXAS contributes to TXA2 biosynthesis and generation. In our study, the baseline expression of TXAS protein is higher in TXAS+/+TP+/+ and TP–/– mice than that in TXAS–/–, and TXAS–/–TP–/– mice (Figure 1). This leads to a higher baseline level of renal TXB2 in TXAS+/+TP+/+ and TP–/– mice than that in TXAS–/– and TXAS–/–TP– /– mice. Because TXAS expression was higher after renal I/R when compared to the control stage, our data confirmed that renal effluent TXB2 concentrations were elevated during the post-reperfusion stage in TXAS+/+TP+/+ and TP–/– mice, but not increased in TXAS–/– and TXAS–/–TP–/– mice. We evidenced that depleting TXAS gene blocks TXAS expression to synthesize TXA2 and produce TXB2, however, deleting TP gene has no effect on the baseline level and post-reperfusion of TXB2 production. Renal warm ischemic injury is associated with a progressive fall in the ratio of vasodilator-tovasoconstrictor eicosanoids during early reperfusion period [39] leading to severely depressed renal microcirculation. This suggests that a decrease in vasodilatory prostacyclin (PGI2) and an increase in vasoconstrictor TXA2 (a decrease in PGI2/TXA2) may appear after renal I/R, however, it needs to be determined in future. Renal injury after suprarenal aortic clamping and reperfusion reduced microvascular renal blood flow and renal function by the loss of ROS mediated medullary and cortical NOS synthesis [40]. Voisin et al. [41] indicated that increased intrarenal NOS expression and a reduction in TXA2 production contribute to a better outcome for the postischemic kidney. It means the vasodilatory NO would counteract vasoconstrictive TXA2 induced the renal hemodynamic downregulation. Our results further showed that blocking TXAS/ TP signaling significantly preserved eNOS expression and renal microcirculation in response to I/R injury. Wang et al. [42] reported that a selective enhanced vasoconstriction of renal afferent arterioles in response to angiotensin II was normalized by a TP antagonist but not by a superoxide dismutase mimetic. Takahashi et al. [43] implicated that the beneficial effects of antioxidant therapy and TXA2 antagonism efficiently reduced preglomerular vasoconstrictor TXA2 and 8-epi-PGF2a depressed glomerular filtration rate and renal plasma flow implicating a protection through TP inhibition. A decrease in TXA2 via COX-1 ameliorates sepsis-induced acute kidney injury [30]. Klausner et al. [44] demonstrated that the vasodilating prostaglandins protect I/R kidney only when vasoconstrictor TXA2 is inhibited. Garvin et al. [21] demonstrated the beneficial effects of TXAS inhibition in reducing renal I/R injury implicating this renoprotection coming from late vasodilatory prostanoid synthesis. Our data showed that gene deletion of TXAS and TP significantly recovered the renal microcirculation after I/R injury with a shorter blood flow recovery time as compared to wild-type mice. This could be in part due to a preservation of eNOS-mediated vasodilatory NO activity and an inhibition of vasoconstrictor TXA2 effect in the gene targeting mice.
The increased TXA2 and isoprostane level by renal I/R injury could activate TP evoking the imbalance of vasodilation and vasoconstriction and leading to severe vasoconstriction in the kidney. In the clinical observations, defective production of TXA2 has a bleeding disorder, whereas overproduction of TXA2 is associated with diverse vascular events including acute coronary syndrome, ischemic stroke, pulmonary hypertension, abnormal renal hemodynamics, and preeclampsia [1,10-13]. Our data evidence the finding that renal I/R increased vasoconstrictor release including TXB2 and ROS and decreased vasodilator NO level by the reduced eNOS expression in the damaged kidney. Platelets from TXAS–/– mice failed to aggregate/generate TXB2 and induce shock and death in response to arachidonic acid as compared to wild-type mice [27]. In another type of sepsis-induced acute renal failure, the degree of renal vasoconstriction and renal blood flow/glomerular filtration rate was significantly decreased in TP–/– mice and TP antagonist-treated mice as compared to wild type mice [2]. Suppression of TXA2 biosynthesis (TXAS inhibition) and combination of TP inhibition confer further antiatherogenic effect in reduction of vascular inflammation, a decrease in macrophages, and an increase in the content of collagen and smooth muscle cells of the atherosclerotic lesions [45]. In a recent study [4], myocardial I/R significantly increased levels of myocardial TXAS, TP, NOx4, IL-1β, apoptosis, coronary endothelin-1, TXB2, O2-. release and the infarct size, with concomitant decreases in eNOS, NO concentrations and cardiac microcirculation, however, these effects were efficiently inhibited in TXAS–/–, TP–/–, and TXAS–/–TP– /– mice. Further, aspirin treatment or knockout of the TXAS, TP or TXAS/TP gene greatly depressed the exaggerated vascular reactivity by vasoconstrictors and vasodilators and efficiently reduced platelet adhesion to the mesenteric endothelium under FeCl3 stimulation [4]. These data implicate the gene targeting on TXAS or TP provides cardioprotection against myocardial I/R injury. In the present study, we further evidenced the attenuation of detrimental effects of enhanced TXAS/TXA2/TP signaling in renal I/R injury by the TXAS– /–, TP–/–, and TXAS–/–TP–/– mice.
Conclusion
Taken together, our results indicate that renal ischemia/ reperfusion activates TXA2 pathway including the enhancement in TXAS and TP protein expression associated with the enhanced inflammation, oxidative stress (apoptosis, autophagy and pyroptosis) and depressed eNOS expression leading to kidney dysfunction. Inhibition of TXAS and TP through TXAS or TP depletion confers renoprotection against ischemia/reperfusion induced oxidative stress and dysfunction. Screening and synthesizing the agents with the combination of TXAS and TP gene deletion like action may provide a therapeutic and preventive strategy to ameliorate renal ischemia/ reperfusion injury.
Highlights
• We established an animal model of transgenic KO mice to determine the role of TXA2 on renal dysfunction in ischemia/ reperfusion injury.
• Blocking TXAS/TXA2/TP signaling through gene targeting provided renal microvascular protection via the restoration of eNOS and renal blood flow in ischemia/reperfusion injury.
• Blockade of TXAS/TXA2/TP signaling confers renal tubular protection against ischemia/reperfusion injury through the action of anti-oxidation, anti-inflammation, anti-apoptosis, anti-autophagy and anti-pyroptosis.
• This study is the first of its kind using transgenic KO mice to explore the role of the TXAS/TXA2/TP signaling pathway in microvascular dysfunction. A schematic summary is illustrated in Figure 9.
Acknowledgment
This work was supported by grant MOST-102-2320-B-003-001- MY3 (Dr. Chiang-Ting Chien), and a research fund from Far-Eastern Memorial Hospital (Dr. Kuo-Hsin Chen).
References
- Hirata M. Cloning and expression of cDNA for a human thromboxane A2 receptor. Nature. 1991; 349: 617-620.
- Boffa JJ. Thromboxane receptor mediates renal vasoconstriction and contributes to acute renal failure in endotoxemic mice. J Am Soc Nephrol. 2004; 15: 2358-2365.
- Sakariassen KS, Alberts P, Fontana P, Mann J, Bounameaux H, Sorensen AS. Effect of pharmaceutical interventions targeting thromboxane receptors and thromboxane synthase in cardiovascular and renal diseases. Future Cardiol. 2009; 5: 479-493.
- Chiang CY, Chien CY, Qiou WY, Chang C, Yu IS, Chang PY, et al., Genetic Depletion of Thromboxane A2/Thromboxane-Prostanoid Receptor Signalling Prevents Microvascular Dysfunction in Ischaemia/Reperfusion Injury. Thromb Haemost. 2018; 118: 982-1996.
- Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994; 46: 205-229.
- Catella F, Healy D, Lawson JA, Fitz Gerald GA. 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc Natl Acad Sci USA. 1986; 83: 5861-5865.
- Reilly MP, Delanty N, Roy L, Rokach J, Callaghan PO, Crean P, et al. Increased formation of the isoprostanes IPF2alpha-I and 8-epi-prostaglandin F2alpha in acute coronary angioplasty: evidence for oxidant stress during coronary reperfusion in humans. Circulation, 1997; 96: 3314-3320.
- McAdam BF, Mardini IA, Habib A, Burke A, Lawson JA, Kapoor S, et al. Effect of regulated expression of human cyclooxygenase isoforms on eicosanoid and isoeicosanoid production in inflammation. J Clin Invest, 2000; 105: 1473- 1482.
- Audoly LP, Rocca B, Fabre JE, Koller BH, Thomas D, Loeb AL, et al. Cardiovascular responses to the isoprostanes iPF(2alpha)-III and iPE(2)-III are mediated via the thromboxane A(2) receptor in vivo. Circulation. 2000; 101: 2833-2840.
- Stahl RA, Thaiss F, Wenzel U, Schoeppe W, Helmchen U. A rat model of progressive chronic glomerular sclerosis: the role of thromboxane inhibition. J Am Soc Nephrol. 1992; 2: 1568-1577.
- Dogne JM, Hanson J, Pratico D. Thromboxane, prostacyclin and isoprostanes: therapeutic targets in atherogenesis. Trends Pharmacol Sci. 2005; 26: 639-644.
- Vagnes OB, Iversen BM, Arendshorst WJ. Short-term ANG II produces renal vasoconstriction independent of TP receptor activation and TxA2/isoprostane production. Am J Physiol Renal Physiol. 2007. 293: 860-867.
- Brien M, Larose J, Greffard K, Julien P, Bilodeau JF. Increased placental phospholipase A2 gene expression and free F2-isoprostane levels in response to oxidative stress in preeclampsia. Placenta. 2017; 55: 54-62.
- Cyrus T, Ding T, Pratico D. Expression of thromboxane synthase, prostacyclin synthase and thromboxane receptor in atherosclerotic lesions: correlation with plaque composition. Atherosclerosis. 2010; 208: 376-381.
- Zhang M, Song P, Xu J, Zou MH. Activation of NAD(P)H oxidases by thromboxane A2 receptor uncouples endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2011; 31: 125-132.
- Chien CT, Lee PH, Chen CF, Ma MC, Lai MK, Hsu SM. De novo demonstration and co-localization of free-radical production and apoptosis formation in rat kidney subjected to ischemia/reperfusion. J Am Soc Nephrol. 2001; 12: 973- 982.
- Chien CT, Chang TC, Tsai CY, Shyue SK, Lai MK. Adenovirus-mediated bcl- 2 gene transfer inhibits renal ischemia/reperfusion induced tubular oxidative stress and apoptosis. Am J Transplant. 2005; 5: 1194-1203.
- Yang CC, Yao CA, Yang JC, Chien CT. Sialic acid rescues repurified lipopolysaccharide-induced acute renal failure via inhibiting TLR4/PKC/ gp91-mediated endoplasmic reticulum stress, apoptosis, autophagy, and pyroptosis signaling. Toxicol Sci. 2014; 141: 155-165.
- Perico N, Benigni A, Zoja C, Delaini F, Remuzzi G. Functional significance of exaggerated renal thromboxane A2 synthesis induced by cyclosporin A. Am J Physiol. 1986; 251: 581-587.
- Bresnahan BA, Dufek S, Wu S, Lianos EA. Changes in glomerular thromboxane A2 receptor expression and ligand binding following immune injury. Kidney Int. 1999; 55: 139-147.
- Garvin PJ, Niehoff ML, Robinson SM, Heisler T, Salinas-Madrigal L, Contis J, et al. Evaluation of the thromboxane A2 synthetase inhibitor OKY-046 in a warm ischemia-reperfusion rat model. Transplantation. 1996; 61: 1429-1434.
- Drouin A, Farhat N, Bolduc V, Thorin-Trescases N, Gillis MA, Villeneuve L, et al. Up-regulation of thromboxane A2 impairs cerebrovascular eNOS function in aging atherosclerotic mice. Pflugers Arch. 2011; 462: 371-383.
- Basili S, Pignatelli P, Tanzilli G, Mangieri E, Carnevale R, Nocella C, et al. Anoxia-reoxygenation enhances platelet thromboxane A2 production via reactive oxygen species-generated NOX2: effect in patients undergoing elective percutaneous coronary intervention. Arterioscler Thromb Vasc Biol. 2011; 31: 1766-1771.
- Oates JC, Halushka PV, Hutchison FN, Ruiz P, Gilkeson GS. Selective cyclooxygenase-2 inhibitor suppresses renal thromboxane production but not proliferative lesions in the MRL/lpr murine model of lupus nephritis. Am J Med Sci. 2011; 341: 101-105.
- Massberg S, Brand K, Grüner S, Page S, Müller E, Müller I, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002; 196: 887-896.
- Hur M, Koo CH, Lee HC, Park SK, Kim M, Kim WH, et al. Preoperative aspirin use and acute kidney injury after cardiacsurgery: A propensityscore matched observational study. PLoS One. 2017; 12: 0177201.
- Yu IS, Lin SR, Huang CC, Tseng HY, Huang PH, Shi GY, et al. TXAS-deleted mice exhibit normal thrombopoiesis, defective hemostasis, and resistance to arachidonate-induced death. Blood. 2004; 104: 135-142.
- Patrono C. Biosynthesis and pharmacological modulation of thromboxane in humans. Circulation. 1990; 81: 112-115.
- Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005; 73: 1907- 1916.
- Mederle K, Meurer M, Castrop H, Höcherl K. Inhibition of COX-1 attenuates the formation of thromboxane A2 and ameliorates the acute decrease in glomerular filtration rate in endotoxemic mice. Am J Physiol Renal Physiol. 2015; 309: 332-340.
- Wagner M, Cadetg P, Ruf R, Mazzucchelli L, Ferrari P, Redaelli CA. Heme oxygenase-1 attenuates ischemia/reperfusion-induced apoptosis and improves survival in rat renal allografts. Kidney Int. 2003; 63: 1564-1573.
- Asano K, Taniguchi S, Nakao A, Maruyama T, Watanabe T, Kurokawa K. Distribution of thromboxane A2 receptor mRNA along the mouse nephron segments. Biochem Biophys Res Commun. 1996; 226: 613-617.
- Katugampola SD, Davenport AP. Thromboxane receptor density is increased in human cardiovascular disease with evidence for inhibition at therapeutic concentrations by the AT(1) receptor antagonist losartan. Br J Pharmacol. 2001; 134: 1385-1392.
- Moss NG, Vogel PA, Kopple TE, Arendshorst WJ. Thromboxane-induced renal vasoconstriction is mediated by the ADP-ribosyl cyclase CD38 and superoxide anion. Am J Physiol Renal Physiol. 2013; 305: 830-838.
- Lee DH, Cho HJ, Kang HY, Rhee MH, Park HJ. Total saponin from korean red ginseng inhibits thromboxane A2 production associated microsomal enzyme activity in platelets. J Ginseng Res. 2012; 36: 40-46.
- Pignatelli P, Carnevale R, Di Santo S, Bartimoccia S, Sanguigni V, Lenti L, et al. Inherited human gp91phox deficiency is associated with impaired isoprostane formation and platelet dysfunction. Arterioscler Thromb Vasc Biol. 2011; 31: 423-434.
- Rong S, Hueper K, Kirsch T, Greite R, Klemann C, Mengel M, et al. Renal PKC-epsilon deficiency attenuates acute kidney injury and ischemic allograft injury via TNF-alpha-dependent inhibition of apoptosis and inflammation. Am J Physiol Renal Physiol. 2014; 307: 718-726.
- Thomas DW, Mannon RB, Mannon PJ, Latour A, Oliver JA, Hoffman M, et al. Coagulation defects and altered hemodynamic responses in mice lacking receptors for thromboxane A2. J Clin Invest. 1998; 102: 1994-2001.
- Weight SC, Waller JR, Bradley V, Whiting PH, Nicholson ML. Interaction of eicosanoids and nitric oxide in renal reperfusion injury. Transplantation. 2001; 72: 614-619.
- Myers SI, Wang L, Liu F, Bartula LL. Oxygen-radical regulation of renal blood flow following suprarenal aortic clamping. J Vasc Surg. 2006; 43: 577-586.
- Voisin V, Declèves AE, Hubert V, Colombaro V, Giordano L, Habsch I, et al. Protection of Wistar-Furth rats against postischaemic acute renal injury: role for nitric oxide and thromboxane? Clin Exp Pharmacol Physiol. 2014; 41: 911-920.
- Wang D, Chabrashvili T, Wilcox CS. Enhanced contractility of renal afferent arterioles from angiotensin-infused rabbits: roles of oxidative stress, thromboxane prostanoid receptors, and endothelium. Circ Res. 2004; 94: 1436-1442.
- Takahashi K, Nammour TM, Fukunaga M, Ebert J, Morrow JD, Roberts LJ, et al. Glomerular actions of a free radical-generated novel prostaglandin, 8-epiprostaglandin F2 alpha, in the rat. Evidence for interaction with thromboxane A2 receptors. J Clin Invest. 1992; 90: 136-141.
- Klausner JM, Paterson IS, Kobzik L, Rodzen C, Valeri CR, Shepro D, et al. Vasodilating prostaglandins attenuate ischemic renal injury only if thromboxane is inhibited. Ann Surg. 1989; 209: 219-224.
- Cyrus T, Yao Y, Ding T, Dogné JM, Praticò D. Thromboxane receptor blockade improves the antiatherogenic effect of thromboxane A2 suppression in LDLR KO mice. Blood. 2007; 109: 3291-3296.