
Review Article
Thromb Haemost Res. 2019; 3(3): 1032.
European Clinical, Laboratory and Molecular (2020 ECLM) Diagnostic Work-Up and Classification of Von Willebrand Disease from the Perspectives of Clinicians and Scientists
Smejkal P1*, Jacques Michiels J1,3*, Zapletal O2, Blatny J2, Batorova A4, Pricangova T4, Budde U5, Hermans C6, Mayger K7, Moore G7, Vangenegten I8, Gadisseur A8 and Penka M1
1Department of Clinical Haematology, Masaryk University, Czech Republic
2Department of Paediatric Haematology, University Hospital Brno and Masaryk University, Czech Republic
3Goodheart Institute & Foundation in Nature Medicine, Blood, Coagulation & Vascular Medicine Science Center, The Netherlands.
4Department of Hemostasis and Thrombosis, Medical School of Comenius University Bratislava, Slovakia
5Central Laboratory, Asklepios Kiniken, Germany
6Hemostasis Thrombosis Center, St Luc University Hospital, Belgium
7Department of Hemostasis & Thrombosis, NHS Foundation Trust London, UK
8Department of Hematology, University Hospital Antwerp, Belgium
*Corresponding author: Petr Smejkal, Department of Clinical Haematology, University Hospital and Department of Laboratory Methods, Faculty of Medicine, Masaryk University, Brno Czech Republic
Jan Jacques Michiels, Goodheart Institute & Foundation in Nature Medicine, Blood, Coagulation & Vascular Medicine Science Center, Rotterdam, The Netherlands
Received: September 17, 2019; Accepted: October 23, 2019; Published: October 30, 2019
Abstract
Introduction: The International Society of Thrombosis Haemostasis (ISTH) classification separated Von Willebrand Disease (VWD) into type 1 and type 2 by the use of four insensitive Von Willebrand Factor (VWF) assays Ristocetine Co-factor (VWF:RCo), VWF Antigen (VWF:Ag), Ristocetine Induced Platelet Agglutination (RIPA) and VWF multimers in a low resolution gel. A complete set of VWF parameters is mandatory to discriminate between all variants of VWD type 1, 2 and 3 and includes Bleeding Time (BT), PFA-100 closure times, FVIII:C, VWF:RCo activity, VWF Collagen Binding (VWF:CB), RIPA, VWF propeptide (VWF:pp), multimeric analysis of VWF and the response of FVIII:C and VWF parameters to DDAVP. We here translate the ISTH into European, Clinical, Laboratory and Molecular (2020 ECLM) classification of the Von Willebrand Disease (VWD) related to the domain location of the Molecular (M) defect in the VWF gene to detect all variants of VWD.
ECLM classification: Recessive VWD type 3 (Pseudo-hemophilia) caused by homozygosity or double heterozygosity for non-sense mutations in the VWF gene differs from severe recessive VWD type 1 due to homozygosity or double heterozygosity for a nonsense/missense or double missense. Recessive “type 1” VWD is featured by very low detectable VWF:Ag between 0.01 and 0.05 U/mL and FVIII:C levels between 0.05 and 0.40 U/ml. Obligate carriers of nonsense or missense mutation usually have mild type 1 VWD with normal VWF multimers with variable penetrance of mild bleeding manifestations in particular when associated with blood group O. Recessive type 2C is a secretion/ multimerization defect due to homozygous or double heterozygous gene defects in the D2 domain. Obligate carriers of VWD type 2C may have mild VWF:RCo deficiency, mild bleeding and abnormal 2C like multimer defect in high resolution gel. Recessive VWD type 2 Normandy (N) due to FVIII binding defect due to mutations in the D’D3 domain of the VWF is featured by low FVIII:C, normal or moderate VWF deficiency type 1 VWD and normal VWF multimers.
Dominant VWD type 1/2E is quantitative/qualitative multimerization defect caused by a heterozygous cysteine mutation in D3 domain of the VWF gene resulting in a secretion and clearance defect of VWF not due to proteolysis of the mutant VWF protein. Dominant VWD Vicenza is a rapid clearance defect due to the R1205H mutation in the D3 domain labeled as VWD 1C and featured by equally low levels of FVIIIC, VWF:Ag, VWF:RCo, VWF:CB labeled as VWD 1C with the presence of unusually large VWF multimers after exercise and DDAVP. Mild VWD type 1m or 1sm (smeary multimers) due to mutations in the D4 and C1-C6 domains clearly differs from dominant VWD type 1/2E and VWD 2M or 2U.
Proteolysis of VWF is increased in dominant type 2A due to mutations in the A2 domain and 2B VWD due to gain of RIPA mutations in the A1 domain result in the absence of large VWF multimers, increased triplet structure of each band, decreased ratios for VWF:RCo/VWF:Ag and VWF:CB/Ag and prolonged BT. Dominant VWD type 2M and 2U is caused by loss of RIPA function mutations in the A1 domain featured by decreased VWF:RCo, normal VWF:CB associated with normal and/or smeary VWF multimers (VWD 2M) or relative decrease of large VWF multimers mimicking VWD 2A.
Conclusion: Prospective clinical and basic research studies are warranted to link clinical, laboratory and molecular defect using a complete set of FVIII: C and VWF parameters. Good evidence exists that the combined use of rapid Glycoprotein (GP) VWF:GPIbR and VWF:GPIbM assays and VWFpp on top of classical VWF assays significantly improved or even appeared to be superior for the correct ECLM defined diagnosis and classification of the manifold manifestations of VWD type 1 and 2.
Keywords: Von willebrand disease; Von willebrand factor VWF antigen; VWF collagen binding; VWF ristocetine cofactor; VWF multimers; and VWF propeptide; VWF ristocetine induced platelet aggregation; Autosomal recessive; Autosomal dominant; Heredity; Diagnosis; Classification
Introduction
Von Willebrand Factor (VWF) is a multimeric plasma glycoprotein that acts as a carrier for coagulation factor VIII in the plasma and as a mediator of platelet adhesion to subendothelial after vascular injury (Figure 1) [1-7]. A number of distinct functional domains have been identified within the VWF, including regions involved in binding to factor VIII, to platelet receptor Glycopotein Ib (GPIb), to platelet GP IIb-IIIa, to components of extracellular matrix such as collagen, regions involved in multimerization and dimerization of VWF, and finally domains involved proteolysis and clearance of VWF in various type 1 and type 2 von VWD defects (Figure 1, Table 1&2) [4]. In this study, we provide historical background information and translated the ISTH into the European Clinical, Laboratory and Molecular (2020 ECLM) classification of VWD based on personal experiences and critical appraisal of the literature regarding the clinical, laboratory and molecular characterization of patients with congenital VWD type 1, 2 and 3 (Figures 2,3 and 4) [1-14].
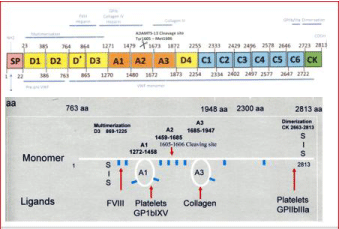
Figure 1: Structure and function of the Von Willebrand Factor (VWF) gene. Pre-pro VWF is cleaved at the Furin Cleavage site 763aa into VWF propeptide (VWFpp
D1D2 domain) and mature VWF monomers (upper). Structure and function of normal Von Willebrand Factor (VWF) protein (lower part).
![]()
- Inherited VWD caused by genetic mutations at the VWF locus includes a broad spectrum of recessive and dominant variants of VWD. Carriers of recessive VWD type 3 or severe recessive type 1 VWD are asymptomatic or may manifest mild bleeding in particular when associated with blood group O.
- Type 1 VWD refers to partial quantitative deficiency of VWF mainly based on a normal VWF:RCo/VWF:Ag ratio above 0.70, type 2 VWD refers to qualitative deficiency of VWF as documented by a decreased VWF:RCo/VWF:Ag ratio below 0.70, and type 3 refers to virtually complete deficiency of VWF.
- Type 2 VWD refers to qualitative variants with decreased platelet dependent function that is attributed to the absence of high molecular weight VWF multimers. The VWF SSC followed a compromised “lumping-spitting” approach in distinguishing four mains categories of type 2A, 2B, 2M, 2N VWD patients.
- Type 2M or 2U is a new entity defined by the SSC-ISTH and refers to qualitative variants with decreased platelet dependent function (VWF:RCo), with the presence of large VWF multimers in a low resolution agarose gel, but recent data show a relative decrease of large VWF multimers in a medium resolution gel.
- Type 2A VWD refers to qualitative variants with absence of HMW multimers, normal or decreased RIPA and decreased VWF:RCo/VWF:Ag ratio below 0.70. By the lumping-splitting approach 2A can be subdivided into the variants of IIA, IIC, IIE and IID as defined by Zimmerman et al (Figure 2).
- Type 2B refers to all qualitative variants with absence of HMW multimers and decreased VWF:RCo/Ag ratio below 0.70 and increased RIPA as first described by� Ruggeri et al (Figure 2).
- Type 2N refers to all qualitative variants with markedly decreased affinity for factor VIII, the presence of large VWF multimers with decreased FVIII:C/VWF:Agratio (less than 0.50) and normal VWF:RCo/VWF:Ag ratio above 0.70.
Table 1: Classification of VWD according to the VWF-SSC-ISTH guidelines 1994-2007 [10-13].
![]()
Classification of Recessive versus Dominant hereditence of VWD
Recessive type 3: pseudohemophilia A of Erikson von Willebrand
VWF:Ag and FVIII: C undetectable or very VWF due double null VWF gene mutations.
Recessive type 1 pseudohemophilia A phenotype due to mutations in the D1 domain
Recessive type 1 and 2C VWD VWF: Ag and VWF:RCo detectable, increased FVIII:C/VWF ratio due to secretion multimerization defect due to mutations in the D1 and D2 domain.
Dominant Type 1: VWF:Ag< 0.30 U/mL
Normal VWF: CB/VWF: Ag and VWF:RCo/VWF:Ag ratio above 0.70
Low VWF type 1: VWF:Ag levels >0.30-0.60 U/mL .
Normal VWF: CB/VWF: Ag and VWF:RCo/VWF:Ag ratio above 0.70
Classification of congenital VWD related to domain located mutations in the VWF gene
Recessive von Willebrand Disease: VWD
Domain
Recessive severe type 3 double null mutation VWF gene double null
Recessive severe or pronounced type 1, homozygous or double missense
D1 D2 D’ D4
Recessive severe type 1 VWD-pseudohemophilia A mimicking type 3
D1
Recessive 2N FVIII: c/VWF:Ag ratio <0.5. FVIII-VWF binding defect
D’-D3
Recessive 2C FVIII: C.VWF: Ag increased, secretion mulimerization defect
D2
Dominant von Willebrand Disease VWD
Domain
2E: type 1/2, loss of large multimers, no triplets and increased clearance
D3
2A: Loss of large MM due to increased VWF proteolysis, RIPA N or decreased
A2
2M: Decreased RIPA, VWF:RCo/VWF:Ag ratio, normal VWF:CB/VWF:Ag ratio
A1
2B: Increased RIPA (0.8mg/ml) and thrombocytopenia with VWD type 2
A1
2CB Collagen binding defect, RCo/VWF:Ag normal and CB/VWF:Ag ratio < 0.7
A3
2D: Dimerization defect, loss of large MM, intervening bands and absence of triplets
CK
Dominant 1sm, dominant 1m or 1 sm with normal (m) or smeary (sm) MM
D4 C1-C6
Table 2: European Clinical Laboratory and Molecular (2020 ECLM) Classification of Von Willebrand Disease (VWD) related to functional Von Willebrand Factor (VWF) domain location [22,23,26,27].
Translation ISTH defined VWD type IIA, IIB, IIC, IIE and IID into ECLM defined VWD 2A, 2B, 2C, 2E and 2D related to domain location of mutations in the VWF gene
In 1980 Ruggeri et al discovered a heightened interaction between platelets and functional abnormal FVIII/VWF protein in a new subtype of type II VWD with increased RIPA and labelled it type IIB. VWD type II, in which the RIPA was decreased or absent, were classified as type IIA [7]. Zimmerman et al. [8] demonstrated in 1986 that proteolysis of VWF is a normal event in normal individuals, increased in type IIA (2A) and IIB (2B) VWD with pronounced triplet structure of each band as the result of increased proteolysis of large VWF multimers (Figure 2). In contrast, proteolysis of VWF is minimal in type IIC (2C), IID (2D) and IIE (2E) variants with aberrant multimeric structure of individual oligomers (Figure 2) [4,8,9]. In type IIA (2A) and IIB (2B) the proportion of 176 kDA and 140 kDA fragments were increased related to the intact 225 kDA subunit, and these degraded VWF fragment were not detected or present in only trace amount in types IIC (2C), IID (2D) and IIE (2E) VWD (Figures 2) [4,8,9]. The normal multimeric pattern (N) in plasma from normal controls displays a typical triplex structure with the presence of the high molecular weight multimers (large MM). Budde & Schneppenheim nicely confirmed the different mechanisms to explain the aberrant structure of individual oligomers in type IIA, IIB, IIC, IID and IIE VWD patients (Figure 3) [4,9].
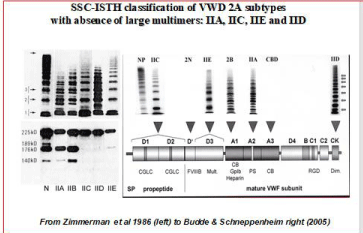
Figure 2: The Zimmermann and SSC-ISTH Classifications of VWD subtypes IIA, IIC, IIE and IID and 2N [10-12].
The classification of congenital Von Willebrand Disease (VWD) in the literature is much too much dominated by the recommendations of the VWF Scientific Standardization Committee (VWF-SSC) at annual SSC meetings of the International Society on Thrombosis and Haemostasis (ISTH) between 1993 and 2007 [10-12]. The VWF-SSC classification of VWD patients in Table 2 is based on a few rather insensitive laboratory tests including FVIII:C, VWF:Ag, VWF:RCo, Ristocetin Induced Platelet Aggregation (RIPA) and VWF multimers in low resolution gel, and therefore cannot clearly distinct between the various variants of type 1 from type 2N, 2M or 2E VWD at VWF levels below 0.15U/ml [1,2]. Type 1 VWD refers to partial quantitative deficiency of VWF mainly based on a normal VWF:RCo/ VWF:Ag ratio, type 2 VWD refers to qualitative deficiency of VWF as documented by a decreased VWF:RCo/VWF:Ag ratio, and type 3 refers to virtually complete deficiency of VWF (Table 1). Type 2 VWD refers to qualitative variants with decreased platelet dependent function that is attributed to the absence of high molecular weight VWF multimers (Table 1). The VWF SSC of the ISTH followed a compromised “lumping-spitting” approach in distinguishing four mains categories of type 2A, 2B, 2M, 2N VWD patients when the Budde, Schneppenheim & Ruggeri classification is applied (Figure 3). Type 2A VWD refers to qualitative variants with absence of HMW multimers, normal or decreased RIPA and decreased VWF:RCo/ Ag ratio. By the lumping-splitting approach 2A can be subdivided in the clearly defied variants of IIA, IIC, IIE and IID as defined by Zimmerman et al. (Figure 2). Type 2B refers to all qualitative variants with absence of HMW multimers and decreased VWF:RCo/VWF:Ag ratio and increased RIPA as first described by Ruggeri et al. VWD type 2M or 2U is a poorly defined entity defined by the SSC-ISTH and refers to qualitative variants with decreased platelet dependent function (VWF:RCo), with the presence of large VWF multimers in a low resolution agarose gel, but recent data show a relative decrease of large VWF multimers in a medium resolution gel (Figure 3 and 4). VWD type 2N (Normandy) refers to all qualitative variants with markedly decreased affinity for factor VIII, the presence of large VWF multimers and normal VWF:RCo/VWF:Ag ratio as first discovered by Mazurier et al.
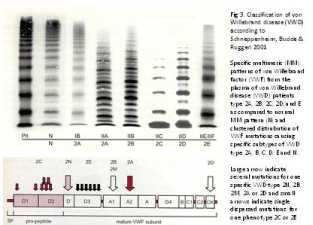
Figure 3: Molecular classification of von Willebrand Disease type 2 related to VWF domain location according to Schneppenheim, Budde and Ruggeri 2001 [4].
For reasons of simplicity, the SSC-VWD of the ISTH used a “lumping” for the classification of type 2 VWD not based on structure and function relationship of VWF gene mutations and functional domains of the VWF protein [10-12]. In the “lumping” approach, type 2 is independent of multimer structure and refers to all loss-offunction variants including FVIII binding defect (FVIII:B defect = Normandy = 2N), loss of High Molecular Weight (HMW) multimers with decreased Ristocetin Induced Platelet Aggregation (RIPA) as 2A thereby lumping IIA, IIC, IID as 2A (Figure 2) or loss of HMW and increased RIPA (2B). Lumping is easier to apply at the expense of illogical grouping of several variants of VWD IIA, IIC, IID, IIE as 2A with decreased RIPA and loss of large VWF multimers due to various mechanisms: increased proteolysis in VWD type 2A and 2B, multimerization defect in VWD 2E, and dimerization defect in VWD 2D. According to Michiels et al. [13] VWD 2M and 2U is featured by decreased or absent RIPA due to loss of function in the interaction of platelet-GPIb-VWF and is associated with normal or smeary VWF multimers or relative loss of large multimers (Figure 3) [1,2,5]. VWD type Vicenza has “supranormal” VWF multimers has been labeled in the past as 2M and changed into VWD type 1C phenotype due to increased clearance. VWD Normandy is labeled as 2N, but has normal VWF multimers and shows a typical mild hemophilia A with normal or mild type 1 VWD phenotype when caused by a noncystein mutation in the D’ domain of the VWF gene.
In the “spitting” alternative according to the ECLM criteria, type 2 VWD refers to the loss-of-function (VWF:RCo) attributed to the absence of HMW multimers separating 2A, 2C, 2E and 2D with decreased RIPA and 2B with increased RIPA (2B) (Figure 4) [1,2]. VWD type 2M according to Michiels [13] is type Ib according to Budde [4] is frequently labeled according to the ISTH classification as 2U, 2A-like or variant 2A. The common feature VWD 2M, 2U and variant 2A is the loss of RIPA function (VWF:RCo and RIPA) due to mutations in the A1 domain. Diagnosis of so-called severe type 1 VWD with VWF:Ag and VWF:RCo or VWF:CB levels below 0.15U/L using the ISTH criteria in Table 1 includes various variant of type 1 secretion and/or clearance defects as well as VWD type 2M [2]. The original description of dominant VWD type 2E by Zimmerman et al. (Figure 2) has a laboratory phenotype 1E or 2E (Figure 4) [8]. Diagnostic differentiation of so-called severe type 1 VWD using the ISTH criteria in Table 1 remains a persistent problem in routine daily practice, which can easily overcome by a correct interpretation of VWF-FVIII:C response curves to DDAVP [1,2,13,14].
Bleeding score assessment
Bleeding severity in patients with VWD has been clinically graded by Eikenboom et al. (1993) [10] and modified by Michiels et al. (2002) [13] as very mild, mild, moderate and severe:
Very mild: the patient has only one or two unclear minor bleeding symptoms and no bleeding history in childhood.
Mild: the patient has one or two obvious bleeding symptoms such as recurrent epistaxis, profuse menstruation, or frequent subcutaneous hematomas (bruising), which usually do not require treatment. This is usually mild type 1 VWD.
Moderate: the patient has more than two bleeding symptoms and has needed a FVIII:C-VWF concentrate transfusion because of abnormal bleeding after an operation or trauma, or both, or has bled for more than 24 hours after tooth extraction. A moderate bleeding type is usually recognized in childhood in type 2 VWD and in pronounced type 1 VWD.
Severe: The patient has pseudo-hemophilia, hemarthrosis, muscle bleeding, and a need for prophylactic treatment with FVIII:C-VWF or VWF concentrate. This is usually type 3 VWD.
On top of clinical bleeding severity assesment we determine grading of bleeding symptoms by use of the modified Adult and Pediatric Vicenza VWD Bleeding Questionnaire and Scoring System (Table 3). Each symptom was described using scale from -1 to +4 points. Bleeding Score (BS) greater than 3 points was considered as positive. Comparison of the Brno results with recent literature is presented in Table 4. VWD type 3 patients have the highest bleeding scores. There is a broad range of bleeding scores from normal to rather severe in VWD type 1 and type 2 VWD patients. The median bleeding score in VWD type 1 and type 2 is not significantly different. In one study, but clearly different in two studies very likely reflecting different VWD patient populations. VWD 2A patients have higher bleeding scores than VWD 2B, 2M and 2N patients.
![]()
Symptom
-1
0
1
2
3
4
Final
scoreEpistaxis
-
No or trivial (<5)
>5 OR more
than 10'Consultation only
Packing or cauterization or antifibrinolytics
Blood transfusion or replacement therapy or desmopressin
Cutaneous
-
No or trivial (<1cm)
> lcm AND no trauma
Consultation only
Bleeding from
minor wounds-
No or trivial (<5)
>5 OR more
than 5Consultation only or steri-strips
Surgical hemostasis or antifibrinolytics
Blood transfusion or replacement therapy or desmopressin
Oral Cavity
-
No
Reported at least one
Consultation only
Surgical hemostasis or antifibrinolytics
Blood transfusion or replacement therapy or desmopressin
GI Bleeding
-
No
Identified cause
Consultation or Spontaneous
Surgical hemostasis,
anti fibrin., blood
transf., replacement
therapy,
desmopressinTooth extraction
No bleeding in at least 2 extractions
None done or no bleeding in 1
Reported, no consultation
Consultation only
Resuturing, repacking or antifibrinolytics
Blood transfusion or Replacement therapy or Desmopressin
Surgery
No bleeding in at least 2 surgeries
None done or no bleeding in 1
Reported, no consultation
Consultation only
Surgical hemostasis or antifibrinolytics
Blood transfusion or Replacement therapy or Desmopressin
Menorrhagia
-
No
Reported, or consultation on ly
Antifibrinolytics or Pill use
D&C, Iron therapy
Blood transfusion or replacement therapy or desmopressin or hysterectomy
Post-partum
hemorrhageNo bleeding in at least 2 deliveries
No deliveries or no bleeding in 1 delivery
Reported, or consultation on ly
D&C, Iron therapy Antifibrinolytics
Blood transfusion or Replacement therapy or Desmopressin
hysterectomy
Muscle hematomas
-
Never
Post traumano therapy
Spontaneous, no therapy
Spontaneous or traumatic, requiring desmopressin or replacement therapy
Spontaneous or traumatic, requiring surgical intervention or blood transfusion
Hemarthrosis
-
Never
Post traumano therapy
Spontaneous, no therapy
Spontaneous or traumatic, requiring desmopressin or replacement therapy
Spontaneous or traumatic, requiring surgical intervention or blood transfusion
CNS Bleeding
-
Never
Subdural, any intervention
Intracerebral, any intervention
Childhood bleeding
For each bleeding:-1
0
No1
Reported2
Consultation
only3
Surgical hemostasis,
antifibrinolytics, iron
therapy4
Blood transfusion or
replacement therapy
or desmopressinUmbilical stump
Cerebral
At circumcision
At venipuncture
Suction bleeding
Hematuria
Menarche
Total Scores:
Table 3: Adult and Childhood Bleeding Score used in the Brno VWD cohort.
![]()
Type of VWD
1
2
3
normal
controlsdefinitive
possible
2A
2E
2B
2M
2N
{3}* Gill 1C. Blood (ASH Annual Meeting Abstracts) 2008; 112: obstruct 425
n
149
17
I
18
4
6
28
234
Q25%-50%
3-10
3-13
10.5-23
-1-1
median
6
7
15
0
{1}* Biss TT, 1 Month Haernost 2010 8: 950-6
n
40
38
6
16
21
range
2-18
0-15
3-17
4-29
-1-2
median
9
2
14
12
0
{2}* Bowmon M,1 Thromb Hoemost 2008; 6; 2062-6
n
16
4
9
1
12
100
0 � 2S0
-3.2-3.6
median
8
16
19
mean 0.16
Department of Clinical Hematology - Department of Pediatric Hemotology, university Hospital Brno
n
53
17
19
2
24
6
range
0-18
1-23
0-16
9-11
0-18
13-25
median
6
10
8
10
4
15
Table 4: Results of the Adult Pedriatic Bleeding Score (BS) in 62 VWD type 2 patients from the Brno cohort compared to three reports in the literature.
Translation of ISTH into 2020 ECLM Classification of VWD
One of the main drawbacks of the ISTH nomenclature and classification that the definitions of VWD type 1, 2 and 3 are based on simple insensitive laboratory parameters and the distinction of normal VWF MM versus abnormal VWF MM missing some or most of the large MM in low resolution gels (Table 1). A molecular approach to the classification of VWD was proposed by, Budde, Schneppenheim & Ruggeri for specific variants of VWD type 2 is based on improved methods of medium and high resolution gels in distinguishing a specific VWF multimeric pattern for each of the main type 2 VWD patients, which is related to a clustered distribution of VWF mutations causing each of the specific subtypes of VWD type 2A, 2B, 2C, 2D 2E and 2N (Figure 3). In VWD Ib [4] (Figure 3) and 2M [13] (Figure 4), there is a relative or no significant decrease of large multimers compared to N, and the triplet structure appears normal but may show a smeary pattern frequently seen in VWD 2M (Figure 4). In VWD type 2A, there is a relative decrease in large VWF multimers compared to normal, and the outer sub-bands of each individual band show markedly pronounced triplets due to increased proteolysis. VWD type 2B cannot be distinguished from VWD type 2A by multimeric analysis alone since their multimeric pattern are similar pronounced due to increased proteolysis, which in VWD type 2B is cause by a gain of function in the platelet GP1b- VWF interaction as reflected by the increased Ristocetin Induced Platelet Aggregation (RIPA) at low concentration of ristocetin. In VWD type 2C (usually autosomal recessive) there is a lack of large VWF multimers and an absence of triplet structure of the individual multimers due to a multimerization/secretion defect in endothelial cells caused by mutations in the VWF propeptide (VWFpp) domains D1 or D2. In VWD type 2D low molecular weight multimers and especially the first band (probably a dimer of VWF) and a tetramer are markedly pronounced with the presence of intervening bands representing multimers with an odd number of monomers are the result of a dimerization mutation defect in the CK domain. In VWD 2E the multimeric pattern is characterized by a lack of or relative decrease of large VWF multimers with the absence of the outer subbands (indicative for absence of increased proteolysis) due rapid clearance of VWF protein due to a multimerization mutation defect in the D3 domain. In VWD 2N, the VWF multimeric pattern is but the VWF protein lacks the capacity to bind FVIII due to mutation in the FVIII binding domain of the VWF gene and protein.
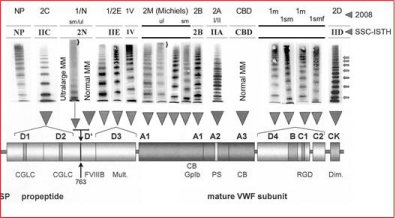
Figure 4: European Clinical, Laboratory and Molecular (ECLM) classification and clustered domain distribution of Von Willebrand Factor (VWF) gene mutations in
Willebrand Disease (VWD) type 1 and type 2 [4,10,49-51,71-73].
Laboratory diagnosis and classification of VWD patients should be based on a complete set of laboratory measurements including bleeding time, PAF-100 closure time, FVIII:C, VWF:Ag, VWF:RCo, VWF:CB, RIPA, the response of VWF and FVIII: C to DDAVP, and analysis of VWF multimeric pattern using low medium and high resolution gels to classify VWD according to established criteria [1,2,10-13]. Three main categories of VWD can be distinguished: first recessive type 3 and severe type 1, second dominant type 1 and 2, and third a large group of mild VWD with no or low penetrance of bleeding manifestations [1,2,13,14]. In general VWD type 1 is a quantitative VWF deficiency with equally decreased values of all VWF parameters (‹0.60 U/ml), a normal ratio for VWF:RCo/VWF:Ag and VWF: CB/ VWF: Ag (›0.60 or ›0.70) before and after DDAVP. VWD type 2 is a qualitative VWF deficiency with normal, near normal or decreased levels for FVIII:C and VWF: Ag and much lower values for VWF:RCo and VWF:CB with deceased ratios for VWF:RCo/VWF:Ag and VWF:CB/VWF:Ag (‹0.60 or 0.70 ISTH). VWF multimeric analysis using low and medium resolution gels clearly differentiates between VWD type 2A, 2C, 2E and 2D (Figures 4). The responses of FVIII and VWF parameter to intravenous DDAVP is an essential tool in the spitting approach, will clearly distinguish pseudo-VWD from true type 1 VWD, will distinguish the various variants of dominant type 1 and 2 (Figure 5), and will elucidate differences between homozygous or compound heterozygous autosomal recessive type 3 and severe type 1 VWD [1,2,5,6,13,14]. The interpretation of VWF response curves to DDAVP has significant therapeutic implications for the different variants of recessive and dominant type 1 and type 2 VWD mutations for both clinicians and VWD patients [13,14]. Responses of FVIII:C and VWF parameters to DDAVP related to the structure and function relationship between laboratory phenotype and expression studies of VWF gene mutations significantly contribute to better understanding of the pathobiology of mutant VWF for the etiology and characterization of the various types of congenital VWD (Figure 5) [4,9].
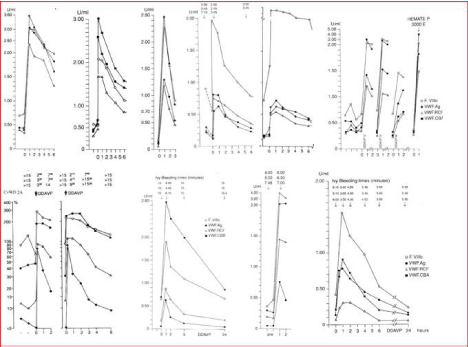
Figure 5: The European Clinical, Laboratory and Molecular (ECLM) VWD classification is based on the ‘splitting’ approach using sensitive and specific VWF
assays, VWF multimeric analysis and response to DDAVP for the detection of a secretion and/or clearance defect of mutated VWF [4,9,13,14,70,71]. The majority
of patients with dominant with pronounced type 1 are Secretion Defect (SD). FVIII-VWF clearance defect VWD 1C is typically seen in Vincenza type VWD 2A, 2B,
2C, 2E and 2M have prolonged Ivy bleeding time (and PFA-CT above 300 seconds) due to decreased functional levels of VWF:CB and VWF: RCo levels usually
below 0.30 U/dl and near normal to normal levels for VWF: Ag.above 0.60 U/mL.
From left to right upper panel. 1: Blood Group O, No VWD; 2: VWD type 1 Secretion Defect (SD) and clearance (C) defect; 3: VWD 1C Vicenza Clearance defect;
4: VWD type 1 SD; 5: VWD type 1 SD; 6: VWD type 1 SD.
From left to right lower panel. 1: VWD 2A with transient correction of VWF parameters after DDAVP; 2: VWD severe 2A with no correction of VWF parameters after
DDAVP; 3: VWD 2B with poor response of VWF: CB and VWF: RCo to DDAVP; 4: VWD 2M. with poor response of VWF:RCO good response of VWF:CB followed
by rapid clearance of FVIII: C and VWF: AF indicating an additional clearance defect.
Autosomal Recessive VWD
VWD Normandy 2N due gene mutations in the factor VIII binding domain (D’-D3, Figures 3 and 4) typically featured by reduced FVIII:C levels despite normal or near normal VWF:Ag, VWF:RCo and VWF:CB levels, decreased FVIII:C/VWF:Ag ratio less than 0.50 and normal VWF:RCo/VWF:Ag ratio together with normal VWF multimeric pattern, normal RIPA and bleeding time mimicking in fact mild hemophilia A. Although some cases of have severe hemophilia A with FVIII:C deficiency of 1U/dL, the majority of 2N VWD patients have mild hemophilia A with FVIII:C levels above 5U/dL [16-20]. Consequently, recessive VWD type Normandy 2N is frequently misclassified as mild hemophilia. Intravenous DDAVP in VWD type Normandy and in mild hemophilia A due to a VWF binding defect of FVIII show completely normal responses for VWF parameters consistent with type 1 VWD, but restricted responses of FVIII:C followed by shortened half-life times [17,19]. Interestingly, homozygous mutations C788R, and C1225G and double heterozygous C788T/null show a hybrid VWD phenotype of pronounced 2E with a factor VIII binding defect (2E/N) (Michiels et al. 2019 manuscript submitted).
The inheritance of VWD type 3 is autosomal recessive [21-26]. Type 3 VWD with virtual complete VWF deficiency are homozygous or compound heterozygous for two null alleles (gene deletions, stop codons, frame shift mutations, splice site mutations, and absence of mRNA) in the majority and rarely compound heterozygous for a null allele and a missense mutation or homozygous for a missense mutation [27]. Compound heterozygosity for a null allele and a missense mutation or homozygosity or double heterozygosity for missense mutations is not consistent with type 3 VWD, but common in patients with severe autosomal recessive type 1 VWD [28-34]. Autosomal recessive severe type 1 VWD patients have detectable but very low VWF levels and FVIII:C levels may range from a few up to around 30% of normal. A considerable number of missense mutations related to autosomal recessive severe type 1 VWD have been identified in the VWFpp D1 and D2 domains and the CK dimerization domain [28-34]. Autosomal recessive VWD type 2C is rare and caused by homozygosity for a missense mutation or double heterozygosity of a null allele and missense mutation in the VWFpp D1 and D2 domains (exon 11 to 16) (Figure 4) [35-41]. Autosomal dominant or recessive VWD 2D is caused by missense mutations in the CK domain (Figure 4).
Autosomal Dominant VWD
The type 2 VWD phenotypes 2A and 2B are clearly different from VWD types 2C, 2D and 2E [35-42]. Careful analysis of reported cases with dominant VWD type 1 due to mutations in the D3 domain (multimerization-secretion-clearance defect) are featured by a type 2E multimeric pattern in medium resolution gels (Figure 4) [42-48]. Budde & Schneppenheim proposed in 2001 and 2005 a novel molecular classification of type 1 and 2 VWD with abnormal multimers (2A, 2B, 2C, 2D, 2E, 2-Normandy versus VWD type 1.2E, 2M or 2U, and type 1sm/2M in Figure 3) mainly based on current knowledge regarding the structure and function relationship of the normal and mutated VWF gene and proteins [4,10,49-52]. By applying a complete set of laboratory tools including a sensitive method for the analysis of the VWF multimeric pattern Budde & Schneppenheim produced consistent good data on the relation between VWF gene mutations and the proposed classification of dominant VWD type 1 and 2 in Figure 3 [4,10,49-52].
Carriers of missense mutations R763Q, R760C, Y795C located in or around the VWFpp Furin cleavage site 763 have ultra-large VWF multimers with a smeary (sm) pattern on top of VWD type Normandy when associated with R854Q or null allele, whereas ultralarge VWF multimers are also seen in VWD Vicenza R1205H/M740I, but such ultra-large VWF multimers are not seen in VWD type 2E (Figure 4) [53-55].
About half of patients diagnosed as type 1 in the MCMDM-1 study had abnormal VWF multimers, which were diagnosed as VWD type 2E in the D3 domain, 2M, 2A-like (2U) in the A1 domain, and type 1 with a smeary pattern of VWF multimers in the D4, B1- 3, C1-2 domain, but were never re-diagnosed as type 2A, 2B, 2C or 2D [49-52]. Heterozygous mutations of the D1, D2 and the site D’ domains usually show a normal VWF multimeric pattern [49,50]. All mutations in the FVIII binding domain show a multimers normal VWF multimeric pattern except a few showing 2E phenotype. VWD patients with mutations in the D3 (multimerization) domain (C1130R, C1130G, W1144G, Y1146C, C1190F)) frequently show a type 1 VWD laboratory phenotype but type 2E pattern with relative loss of large VWF multimers and reduced triplet structure and proteolysis (Figure 4). Loss of function mutations in the A1 domain (L1307P, R1315C, R1315L, R1374C, R1374H, G1415D) show the presence but relative loss of large VWF multimers in medium resolution gels, no increase of triplet structure, and a defect of the VWF:RCo and RIPA consistent with VWD type 2M or 2U [4,10]. Several mutations (L2207P, C2257S, C2304Y, R2379, G2441C, R2464C, R2464C, C2469P, C2671Y) in the D4, B1-3, C1-2 domain show a dominant type 1sm VWD phenotype featured by an atypical smeary pattern of large multimers and no triplet structure of small multimers [49,50].
Data from the European MCMDM-1VWD study show that the heterozygous mutations in the D4, B1-3, and C1-2 domains (L1774S*, K1794E*, C2304Y*, R2313H, G2518S*, Q2544X*, C2693Y*, and P2722A) have normal multimers and have mild VWD type 1m disease with variable penetrance of bleeding manifestations. Heterozygous mutations in the D4, B1-3, C1-2 and CK domains V1822G*, L2207P*, C2257S*, C2304Y*, C2362F*, G2441C*, R2464C*, C2477Y*, C2477S*, and Q2520P* have mild to moderate VWD type 1, abnormal VWF multimers (usually smeary pattern) [51,52]. Nearly all mutations in the D4, B1-3, C1-2 and CK domain have increased FVIII:C/VWF:Ag ratios around or above 2 (indicated by an astrix*) indicating a secretion (S) defect [19], which predict restricted response of VWF to DDAVP and more or less rapid clearance (C) after DDAVP (VWD type 1 SC). Expression studies of mutant VWF are predicted to show abnormal banding of VWF multimers as the cause of a smeary pattern, which is more pronounced after DDAVP [51,52].
The clinical importance of DDAVP response curves to diagnose and classify VWD
Desmopressin (DDAVP, 1-desamino-8-D arginine vasopressin) is a synthetic analogue of the natural hormone vasopressin [56], but it has no pressor activity, in contrast to vasopressin. DDAVP stimulates the endogenous release of endogenous factor VIII (FVIII:C) and Von Willebrand Factor (VWF) proteins. The effect is virtually immediate, usually with 2-6-fold increases in the plasma concentrations of FVIII, VWF:Ag, VWF:RCo and VWF:CB and tissue plasminogen activator [57,58]. Platelet function and coagulation assay, RIPA and the VWF assays VWF:Ag ELISA, VWF:RCo, VWF:CB ELISA assay and VWF multimeric analysis has been described in great detail by Michiels et al in 2002. Ivy bleeding time (BT), PFA-100 closure time, and all FVIII-VWF parameters FVIII:C, VWF:Ag, VWF:RCo, vWF:CB and VWF multimers before and after a single DDAVP challenge test were measured before (t=0) and after 1, 2, 4, and 24 hours postinfusion of DDAVP iv according to the Rotterdam protocol proposed by Dr Manucci on behave of the SCC ISTH (Table 5). The biological characteristics and half-life times of the VWF parameters were determined at 5 time points for the values between 15 minutes and 6 hours after termination of intravenous DDAVP. Between 1992 and 2000 Michiels & Van Vliet of the Blood & Coagulation Laboratory, Department of Hematology Erasmus University Medical Center Rotterdam prospectively evaluated the combined use of FVIII:C, VWF:Ag, VWF:RCo, VWF:CB and responses to DDAVP in VWD type 1 and 2 to improve the shortcomings of the 1994 ISTH classification of VWD patients in recessive type 3 and severe 1, in mild and pronounced dominant type 1 type 2A, 2B and 2M (Figure 5).
Standardization of DDAVP response curves
DDAVP Response Curves (RC) over at least six hours post- DDAVP to study VWF clearance should follow the SSC-ISTH recommendations (Table 5). Candidate election for a diagnostic DDAVP:RC test should be very selective and restricted to the proband of the family with a particular VWD molecular defect, simple because you can predict a similar response to DDAVP in the other family members with the same molecular defect.
The DDAVP trial should start between 8.00 and 9.00 O’clock a.m. and should be performed by a skilled nurse after informed consent of the patient by the responsible physician. Blood sampling up to 6 hours is essential or evaluation of secretion and clearance pattern in VWD type 1 and 2. Blood sampling at 12 and 24 hours is optional but of high importance to differentiate between mild VWD and normal. The responses of the FVIII:C and VWF parameters to intravenous DDAVP is an essential tool in the splitting approach for three reasons:
1. It will clearly distinguish pseudo-VWD from true type 1 VWD.
2. It will distinguish the various variants of dominant type 1 and 2.
3. It will elucidate differences between homozygous or compound heterozygous autosomal recessive type 3 and severe type 1 VWD (Table 6) [1,2,13,14].
![]()
Response to DDAVP: VWD type Mutation
FVIII: C
VWF:Ag
VWF:RCo
VWF:CB
BTcorrection
Mild 1
Variable
Good
Good (G)
Good
Good
Yes
Variable penetrance of bleeding
Rec 2N
D’-D3
Poor
Good
Good
Good
BT is normal
Short
Dominant VWD
1/2E
D3
Good
Transient
Transient
Transient
Transient
1/Vicenza
D3
Short/G
Short/G
Short/G
Short/G
Short/Yes
2M
A1
Good
Restricted
Poor
Restricted
Transient
2B
A1
Good
Good
Poor
Poor
No
2A group I
A2
Good
Restricted
Poor
Poor
No
2A group II
A2
Good
Good
Short/G
Short/G
Transient
1m, sm,smf
D4 B1-3, C1-2
Good
Restricted/G
Restricted /G
Restricted/G
Yes
2D
CK
Partial
Partial
Poor
Poor
No
Table 6: DDAVP response of FVIII:C and VWF parameters to DDAVP in patients with VWD type 1 and type 2 [13,14,59-66].
A normal response of VWF parameters and restricted response of FVIII:C to DDAVP followed by shortened half life times of FVIII:C refers to VWD 2N due to a FVIII binding defect in the VWF or to mild hemophilia due to a VWF binding defect in the FVIII protein [17,19,20]. A short half life time of the VWF parameters and FVIII:C after DDAVP in type 1C Vicenza, 2E, 2M and 2U indicates rapid clearance of VWF antigen not due to proteolysis as demonstrated by the absence of triplet structures of VWF bands and absence of VWF degradation products [13,14]. Short half-life times of functional VWF parameters as compared to near normal to normal half-life time for VWF:Ag in type 2A and 2B indicates increased proteolysis as demonstrated by the loss of large VWF multimers, the presence of VWF degradation products (Figure 3) and increased triplet structure of VWF multimer bands [2,14].
Patients with recessive type 3 do not respond to DDAVP. Severe recessive VWD type 1, 2C and 2D respond poorly or not to DDAVP with regard to the VWF parameters but usually show an increase of FVIII:C to normal. The response to DDAVP in VWD type 1 and 2 is dependent on the severity of the disorder (Table 6). Patients with mild VWD type 1 with normal VWF multimers and VWF values between 0.30 and 0.60 usually respond well to DDAVP (Table 6). Interestingly, the response to DDAVP of FVIII:C is 2 to 3 times higher to that of VWF:Ag in carriers of a null allele or missense mutation (parents of VWD patients with recessive type 3 or severe type 1) [1,51]. The responses of FVIII:C and VWF parameters are of huge importance for the diagnostic work-up severity assessment and characterization of the VWF defect related to domain location of VWD type 1 and type 2 mutations [59-66]. The laboratory phenotype of dominant VWD type 2M or 2U due to loss of GPIb function mutation in the A1 domain is characterized by decreased RIPA in the presence of a near normal VWF multimeric pattern in a low resolution gel, a poor response to DDAVP of VWF:RCo and good responses to DDAVP of both VWF:CB and VWF:Ag and FVIII:C followed by decreased half life times of VWF parameters. This type of response curves to DDAVP is corroborated by increased VWFpp/ Ag ratios in patients with VWD type 2 M as shown e.g. in VWD 2M due to the I1416N mutation [65]. Patients with dominant VWD severe type 2A (group I) have pronounced or very low VWF:RCo, prolonged to very prolonged BT, PFA-100 Closure Times (CT) longer than 250 seconds, and show a good response of FVIII:C and VWF:Ag but a minor or poor response of VWF:RCo to DDAVP with no correction of the BT (Table 3) are therefore candidates for VWFFVIII concentrate substitution for the treatment and prophylaxis of bleeding symptoms. A minority of VWD mild type 2A (group II) are featured by near normal to prolonged values for BT, normal FVIII:C and VWF:Ag, low VWF:RCo and VWF:CB, a normal RIPA and complete correction of BT and functional VWF parameters to normal for only a few hours followed by short half life times for VWF:RCo and VWF:CB (Table 3). These mild type 2A VWD (group II) patients have transiently complete response of FVIII and VWF parameters to DDAVP that may be good enough for the treatment and prophylaxis of spontaneous minor bleedings, but will not be enough for bleeding prophylaxis in surgical and trauma. A good DDAVP response curve (challenge test) can be used as sufficient for the treatment of spontaneous minor bleeds. major bleeds after trauma, for prevention of bleeding in connection with surgery or other invasive procedures, if VWF:RCo and FVIII:C reach normal levels after infusion DDAVP followed by near normal or normal half-life times. If the DDAVP response of VWF parameters is insufficient or the response duration is short, a VWF/FVIII concentrate should be considered [59-66].
The role of FVIII:C/VWF:Ag ratio in the diagnosis and classification of VWD
By definition, the concentration in plasma of FVIII:C and VWF:Ag is 1U/ml [13]. Consequently, the ratio FVIII:C/VWF:Ag is around 1 in normal individuals with blood group O and non-O [15].
A decreased FVIII:C/VWF:Ag ratio of less than 0.5 is caused by FVIII:C binding defect of the VWF protein in recessive homozygous or double heterozygous type 2N VWD
A decreased FVIII:C/VWF:Ag ratio is also seen in mild hemophilia A due to a VWF binding defect in the FVIII gene [16-20]. A normal response of VWF parameters and restricted response of FVIII:C to DDAVP followed by shortened half-life times of FVIII:C refers to VWD 2N due to a FVIII binding defect in the VWF or mild hemophilia due to a VWF binding defect in the FVIII protein [17,19,20].
The ratio of FVIII:C binding sites on VWF:Ag on a molecular basis is 1:50 and independent of the size of VWF multimers indicating that many potential FVIII:C binding sites on VWF:Ag are free. As the VWF:Ag is 50% of normal in quantitative VWD type 1 due to a secretion defect of the mutated VWD protein, the ratio of FVIII:C/ VWF:Ag will increase to about 2 [15]. Carriers of recessive VWD type 3 heterozygous for the VWF null allele (decreased synthesis) have an increased ratio of 2.06 for FVIII:C/VWF:Ag. The increased FVIII/VWF:Ag ratio is clearly related to the severity of the VWF:Ag deficiency VWF due to a synthesis (null allele) or secretion (missense allele) defect with FVIII:C/VWF:Ag ratios of 3.2, 1.96 and 1.46 at VWF:Ag plasma levels of ‹30, between 30-60 and above 60U/ml [15].
An increased FVIII:C/VWF:Ag ratio in VWD type 1 and 2 refers to a VWF synthesis (null allele) or secretion (missense) defect of mutated VWF
A VWF synthesis or secretion defect in carriers of a null allele or missense allele in mild VWD type 1 and 2 as can be documented by a restricted response of VWF;Ag to DDAVP and a normal response of FVIII:C to DDAVP [13]. A poor response of VWF:Ag as compared to a restricted response of FVIII:C to DDAVP results in a pronounced increase of the FVIII:C/VWF;Ag ratio in severe recessive type 1 and recessive type 2C VWD (asymptomatic parents showing mild type 1 VWD with increased FVIII:C/VWF:Ag ratio after DDAVP. A normal FVIII:C/VWF:Ag ratio is consistent with normal secretion of mutant VWF protein in type 1 (including Vicenza) and in type 2 (2A group II and 2B) VWD patients [2,13]. In VWD 2A, rapid proteolysis of large VWF multimers results in rapid loss of functional VWF:RCo and VWF:CB after DDAVP but normal levels of degraded VWF:Ag levels after DDAVP followed by normal or near normal half-life of VWF:Ag and VWF pp/Ag ratios between 1.0 and 2.0.
The role of VWFpp/VWF: Ag ratio in the diagnosis and classification of VWD
normal, whereas the clearance of VWF:Ag may be very short, shortened or normal. VWD patients with a secretion defect but normal clearance and who thus show a restricted response of VWF to DDAVP followed by normal half lifetimes of VWF:Ag are expected to have decreased values for VWFpp and VWF:Ag: both low, ratio 1:1 reflecting the secretion defect in VWD due to stop codon or missense, whereas a normal ratio for VWF:pp/VWF:Ag reflects a normal clearance of VWF:Ag (Figure 5). VWD patients with a normal secretion but increased clearance of VWF:Ag (and associated functions VWF:RCo and FVIII:C) show good responses to DDAVP followed by short half life times of VWF:Ag are expected to have an increased VWF:pp/VWF:Ag ratio (reflecting increased clearance VWF:Ag). Short half-life times of VWF:Ag, VWF:CB, VWF:RCo parameters and FVIII:C after DDAVP in type 1, Vicenza, 2E, 2M and 2U results in increased VWF:pp/VWF:Ag ratios indicates rapid clearance of the VWF:Ag/FVIII complex not due to proteolysis on top of a multimerization defect in VWD 2 E, and loss of RIPA defect in VWD 2M and 2U [13,14]. Short half-life times of functional VWF parameters VWF:RCo and VWF:CB caused by increased proteolysis in type 2A and 2B is associated with near normal or normal VWF:Ag half-life times and normal or slightly increased VWF:pp/VWF:Ag ratios [2,14].
We have critically analysed the literature on the results of VWF:pp/VWF:Ag ratios in relation to the level of VWF:Ag and the VWF:Ag survival times after DDAVP in VWD type 1/2E, type 1 Vicenza due to mutations in the D3 domain, VWD type 1SC due to a mutation in the D4 domain, mild VWD type 1 due to mutations in the D4-B1-B3-C1-C2 domains and mild VWD type related to the C1584 mutation with blood group O (Figure 6) [67-71]. In case of secretion defect both absolute levels of VWF:Ag and VWF:pp are decreased but the VWF:pp/VWF:Ag ratio remains normal or near normal (Figures 5 and 6). In case of normal secretion but increased clearance of VWF;Ag the ratio VWF:pp/VWF:Ag increases depending on the degree of shortened VWF:Ag half-life time, which can nicely be assessed after a DDAVP challenge test [59,60,63,67-71]. In view of literature Vangenechten et al. and Michiels et al recently studied the performance of a complete set of rapid and classical VWF functional and quantitative assays for improved diagnosis of ECLM defined VWD patients [72-75]. The response of FVIII and VWF parameters to DDAVP in VWD Vicenza (R1205H) is normal indicating normal secretion but followed by rapid clearance and very high VWF:pp/ VWF:Ag ratios (Figures 5 and 6) [13,22,58,66,69,72-75]. Patients with VWD type 1/2E due to mutations in the D3 multimerization domain and the mutation S2179F in the D4 domain have increased VWF:pp/ VWF:Ag ratios despite decreased or low absolute values for VWF:pp and shortened VWF:Ag half-life times indicating rapid clearance as a main mechanism for a laboratory phenotype (Figures 5 and 6). Patients with mild VWD type 1 due to mutations in the D1, D2 and D’ domains have decreased VWF:pp levels and normal VWF:pp/ VWF:Ag ratios of just above 1 indicating for a mild secretion defect of VWF (D4-C1-C6 and D1D2 in Figure 1). Patients with mild VWD due to mutations in the D4-B1-B3-C1-C2 domains have decreased values for VWF:pp but slightly elevated VWF:pp/VWF:Ag ratios indicative for the combination of a mild secretion combined with a mild clearance defect (Figure 6). Patients with the C1584 mutation and blood group O have normal values for VWF:pp and near normal VWF:pp/VWF:Ag ratios indicative for a mild clearance defect of VWF (Figure 6) [23]. Patients with recessive severe type 1 VWD are predicted to have decreased values for both VWF:Ag and VWF:pp and normal VWF:pp/VWF:Ag ratios due to a severe secretion defect.
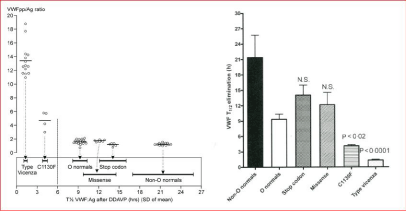
Figure 1: VWF: pp and VWF: Ag related to VWF: Ag half-life times after DDAVP in patients with VWD type Vicenza, 1C, type 1E/ 2E (C1130F) and in mild VWD
type 1 in carriers of a stop codon or missense mutation and in controls with blood group O or non-O. Source Sztkowska & Casonato et al. 2008.
Left. VWF: pp/VWF: Ag ratios in blood group non-O healthy controls, in heterozygous carriers of a stop codon or missense mutation are in the normal range (‹2.0),
slightly increased in blood group O healthy control, increased in C1130F mutated VWD type 2 E and high in R1205H mutated VWD Vicenza type 1C.
Right. Mean VWF: Ag biological half-life times in blood group non-O healthy control versus blood group O healthy control and heterozygous carriers of a stop
codon or missense mutation are in the normal range, but significantly shortened in C1130F mutated VWD type 1 E and very short in R1205 mutated VWD Vicenza
type 1C.
Current recommendations of VWD patient diagnostic work-up and characterization
Recommendations for current future VWD studies cannot be made without knowledge of questions to be posed by the need to better characterize VWD platelets at the clinical, laboratory and molecular defects of specific VWD entities (Figure 7 and Table 2) [70- 72]. The following are recommended tools to be included for clinical and basic research studies.
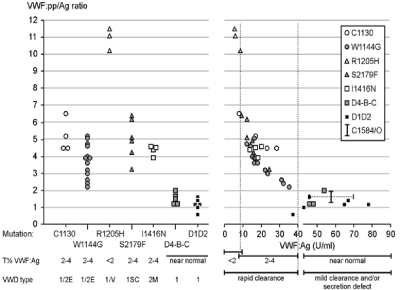
Figure 7: Analysis of VWF: pp/VWF: Ag ratios (Left) related to VWF: Ag half-life times after DDAVP (Right) from two studies on dominant VWD type 1E/2 E and
VWD Vicenza type 1C due to C1130, W1144G, and R1205H mutations in the D3 domain, VWD 2M due to I1416N mutation, and mild VWD type 1 heterozygous
carriers of missense mutation in the D1, D2 and D4-C1-C6. Source Gadisseur et al. 2009 [71].

Figure 8: Standardized DDAVP challenge test over at least six hours proposed in the 1990s by Mannucci on behave of the SSC-ISTH and modified by Michiels et
al in 2002 for the diagnostic work-up and characterization of VWD type 1, 2 and recessive 1 [1].
1. Standardized questionnaire to document clinical severity and quantitate the burden of bleeding in all variants of VWD patients using the original or modified Vicenza Bleeding Score (BS) assessment
2. Samples suitable for plasma, DNA and platelet RNA analysis
3. Plasma phenotype analysis using a complete set of laboratory tools including bleeding time
PFA-100 closure times, FVIII:C, VWF:Ag, VWF:GPIbM, VWF:GPIbR, VWF:pp, VWF:CB (collagen type 1), RIPA and determination of ABO blood group
4. VWF multimer analysis using low, medium or high resolution gels
5. Mutation screening of the VWF promoter and exons 2 to 52
6. Responses of FVIII:C and VWF parameters to DDAVP over at least six hours post-DDAVP in mandatory to study VWF clearance according to the SSC-VWD of the ISTH recommendations (Table 5).
Author’s Contribution
PS and JJM wrote the manuscript on behave of the Antwerp, Brno, London Rotterdam Collaboration and Academic Research on von Willebrand disease: ICAR.VWD.
References
- Michiels JJ, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, van de Velde A, et al. Characterization of recessive severe type 1 and 3 von Willebrand disease (VWD), asymptomatic heterozygous carriers versus blood group O-related von Willebrand factor deficiency and dominant type 1 VWD. Clin Applied Thromb/Hemostas. 2006; 12: 277-295.
- Michiels JJ, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, van de Velde A, et al. Classification and characterization of hereditary types 2A, 2B, 2C, 2D, 2E, 2M, 2N, and 2U (unclassifiable) von Willebrand disease. Clin Applied Thromb/Hemostas. 2006; 12: 397-420.
- Ruggeri ZM. Structure of von Willebrand factor and its function in platelet adhesion and thrombus formation. Best Practice Res Clin Haematol. 2001; 14: 257-279.
- Schneppenheim R, Budde U, Ruggeri ZM. A molecular approach to the classification of von Willebrand disease. Best Practice Res Clin Haematol. 2001; 14: 281-298.
- Michiels JJ, Smejkal P, Penka M, Batorova A, Pricangova T, Budde U, et al. Diagnostic differentiation of von Willebrand types 1 and 2 by von Willebrand Factor Multimer Analysis and DDAVP Challenge test. Clin/Applied Thromb Hemostas. 2017; 23: 518-531.
- Michiels JJ, Batorova A, Pricangova T, Smejkal P, Penka M, Vangenechten I, et al. Changing insights in the diagnosis and classification of autosomal recessive and dominant von Willebrand diseases 1980-2015. World J Hematol. 2016; 5: 61-74.
- Ruggeri ZM, Pareti FI, Mannnucci PM, Ciavarella N, Zimmerman TS. Heightened interaction between platelet and FVIII/von Willebrand factor in a new subtype of von Willebrand disease. N Eng J Med. 1980; 302: 1047-1051.
- Zimmermann ThS, Dent JA, Ruggeri ZM, Nannini LH. Subunit composition of plasma von Willebrand factor. J Clin Invest. 1986; 77: 947-951.
- Schneppenheim R, Budde U. Phenotypic and genotypic diagnosis of von Willebrand disease. Sem Hematol. 2005; 42: 15-28.
- Eikenboom JCJ, Reitsma PH, Peerlinck KJM, Briet E. Recessive inheritance of von Willebrand disease. Lancet. 1993; 341: 982-986.
- Sadler JE, Mannucci PM, Berntorp E, Bochkov N, Boulyjenkov V, Ginsburg D, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemostas. 2000; 84: 160-174.
- Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemostas. 2006; 4: 2103-2114.
- Michiels JJ, van de Velden A, van Vliet HHDM, van der Planken M, Schroyens W, Berneman Z. Response of von Willebrand factor parameters to desmopressin in patients with type 1 and type 2 congenital von Willebrand disease: diagnostic and therapeutic implications. Sem Thromb Hemostas. 2002; 28: 111-131.
- Michiels JJ, van Vliet HHDM, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, et al. Intravenous DDAVP and factor VIII-von Willebrand factor concentrate for the treatment of bleeding in patients with von Willebrand disease type 1, 2 and 3. Clin Applied Thromb/Hemostas. 2007; 13: 14-34.
- Eikenboom JCJ, Castaman G, Kamphuizen PW, Rosendaal FR, Bertina RM. The factor VIII/von Willebrand factor ratio discriminates between reduced synthesis and increased clearance of von Willebrand factor. Throm Haemostas. 2002; 87: 252-257.
- Mazurier C, Goudemand J, Hilbert L, Caron C, Fressinaud E, Myer D. Type 2N von Willebrand disease: clinical manifestations, pathophysiology, laboratory diagnosis and molecular biology. Best Practice Res Clin Haematol. 2001; 14: 337-347.
- Mazurier C, Gaucher C, Jorieux S, Goydemand M. Biological effect of desmopressin in eight patients with type 2N (Normandy) von Willebrand disease. Br J Haematol. 1994; 88: 849-854.
- Caron C, Mazurier C, Goudemand J. Large experience with factor VIII binding assay of plasma von Willebrand factor using commercial reagents. Br J Haematol. 2002; 117: 716-718.
- Jacquemin M, Lavend’homme R, Benhida A, Vanzieleghem B, d’Oiron R, Lavergne JM, et al. A novel cause of mild/moderate haemophilia A: mutations scattered in the factor VIII C1 domain reduce factor binding to von Willebrand factor. Blood. 2000; 96: 958-965.
- d’Orion R, Lavergne JM, Lanend’homme R, Benhida A, Bordet JC, Negrier C, et al. Deletion of alanine in the FVIII C2 domain results in mild hemophilia by impairing FVIII binding to VWF and phospholipids. Blood. 2004; 103: 155- 157.
- Schneppenheim R, Krey S, Bergman F, Bock D, Budde U, Lange M, et al. Genetic heterogeneity of severe von Willebrand disease type III in the German population. Hum genet. 1994; 94: 640-652.
- Zhang ZP, Blomback M, Egeberg N, Falk G, Anvret M. Characterization of the von Willebrand factor gene in von Willebrand disease type III patients from 24 families of Swedish and Finnish origin. Genomics. 1994; 21: 189-193.
- Zhang A, Lindstedt M, Blomback M, Anvret M. Effects of the mutant von Willebrand factor gene in von Willebrand disease. Hum Genet. 1995; 96: 388-394.
- Eikenboom JC, Castaman G, Vos HL, Bertina RM, Rodeghiero F. Characterization of genetic defects in recessive type 1 and type 3von Willebrand patients of Italian origin. Thromb Haemostas. 1998; 79: 709-717.
- Baronciani L, Cozzi G, Cabciani MT, Peyandi F, Scrivastava A, Federici AB, et al. Molecular characterization of a multi-etnic group of 21 patients with type 3 von Willebrand disease. Thromb Haemostas. 2000; 84: 536-540.
- Eikenboom JCJ. Congenital von Willebrand disease type 3: clinical manifestations, pathophysiology and molecular biology. Best Practice Res Clin Haematol. 2001; 14: 365-379.
- ISTH data base of von Willebrand factor mutations: www.shef.ac.uk/vwf
- Allen S, Abuzenadah AM, Hinks J, Blagg JL, Gursel T, Ingerslev J, et al. A novel von Willebrand disease-causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerization and secretion. Blood. 2000; 96: 560-568.
- Castaman G, Lattuada A, Mannucci PM, Rodeghiero F. Factor VIII: C increases after desmopressine in a subgroup of patients with autosomal recessive severe von Willebrand disease. Brit J Haematol. 1995; 89: 147- 151.
- Castaman G, Eikenboom JCJ, Lattuada A, Manucci PM, Rodeghiero F. Heightened proteolysis of the von Willebrand factor subunit in patients with von Willebrand disease hemizygous or homozygous for the C2364F mutation. Brit J Haematol. 2000; 108: 188-190.
- Castaman G, Novella E, Castiglia E, Eikenboom JCJ, Rodeghiero F. A novel family with recessive von Willebrand disease due to compound heterozygosity for a splice site mutation and a missense mutation in the von Willebrand factor gene. Thromb Res. 2002; 105: 135-138.
- Tjernberg P, Castaman G, Vos HL, Bertina RM, Eikenboom JCJ. Homozygous C2362F von Willebrand factor induces intracellular retention of mutant von Willebrand factor in autosomal recessive severe von Willebrand disease. Br J Haematol. 2006; 133: 409-418.
- Castaman G, Bertoncello, Bernardi M, Eikenboom JC, Budde U, Rodeghiero F. Autosomal recessive von Willebrand disease associated with compound heterozygosity for a novel nonsense mutation (2908delC) and the missense mutation C2362F: definite evidence for non-penetrance of the C2363F mutation. Am J Hematol. 2007; 82: 376-380.
- Schneppenheim R, Budde U, Obser T, Brassard J, Mainush K, Ruggeri ZM, et al. Expression and characterization of von Willebrand factor dimerization defects in different types of von Willebrand disease. Blood. 2001; 97: 2059- 2066.
- Ruggeri ZA, Nilsson IM, Lombardi R, Holmberg L, Zimmerman ThS. Aberrant multimeric structure of von Willebrand factor in a new variant of von Willebrand’s disease (type IIC). J Clin Invest. 1982; 70: 1124-1127.
- Holmberg L, Karpman D, Isakson C, Kristofferson AC, Lethagen S, Schneppenheim R. Ins405AsnPro mutation in the von Willebrand factor propeptide in recessive type 2A (IIC) von Willebrand’s disease. Thromb Haemostas. 1998; 79: 718-722.
- Mazurier C, Manucci PM, Parquet-Gernez A, Goudemand, Meyer D. Investigation of a case of subtype IIC von Willebrand disease: characterisation of the variability of this subtype. Amer J Hematol. 1986; 22: 301-311.
- Gaucher C, Diéval J, Mazurier C. Characterization of von Willebrand factor gene defects in two unrelated patients with type IIC von Willebrand disease. Blood. 1994; 84: 1024-1030.
- Battle J, Lopez-Fernandez MF, Lasiera J, Fernandez Villamor AF, Lopez Berges C, Lopez Borrasca A, et al. Von Willebrand disease type IIC with different abnormalities of von Willebrand factor in the same sibship. Amer J Heamatol. 1986; 21: 177-188.
- Battle J, Lopez Gernandez MF, FernandezVillamor A, Lopez Berges C, Zimmerman ThS. Multimeric pattern discrepancy between platelet and plasma von Willebrand factor in type IIC von Willebrand disease. Amer J Hematol. 1986; 22: 87-88.
- Schneppenheim R, Thomas KB, Krey S, Budde U, Jessat U, Sutor AH, et al. Indentification of a candidate missense mutation in a family with von Willebrand disease type IIC. Hum Genet. 1995; 95: 681-686.
- Casana P, Martinez F, Haya S, Espinos C, Aznar JA. Association of the T1156M mutation in the von Willebrand factor gene with dominant type 1 von Willebrand disease. Ann Hematol. 2001; 80: 381-383.
- Lethagen S, Isaksson C, Schaedel C, Holmberg L. Von Willebrand disease caused by compound heterozygosity for a substitution mutation (T1156M) in the D3 domain of the von Willebrand factor and a stop mutation (Q2470X). Thromb Haemostas. 2002; 88: 421-426.
- Eikenboom JCJ, Matsushita T, Reitsma PH, Tuley E, Castaman G, Briët E, et al. Dominant type 1 von Willebrand disease caused by mutated cysteine residues in the D3 domain of von Willebrand factor. Blood. 1996; 88: 2433- 2441.
- Castaman G, Eikenboom JCJ, Missiaglia E, Rodeghiero F. Autosomal dominant type 1 von Willebrand disease due to C1130F mutation in exon 26 of von Willebrand factor gene: description of five families and evidence for a founder effect. Br J Haematol. 2000; 108: 876-879.
- Tjernberg P, Vos HL, Castaman G, Bertina RM, Eikenboom JCJ. Dimerization and multimerization defects of von Willebrand factor due to mutated cysteine residues. J Thromb Haemostas. 2004; 2: 257-265.
- Haberichter SL, balistrreri M, Christopherson P, Morateck P, Gavazova S, Bellissimo DB, et al. Assay of von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006; 108: 3344-3351.
- James PD, O’Brien LA, Hegadorn CA, Notley CRP, Sinclair GD, Huogh C, et al. A novel type 2A von Willebrand factor mutation located at the last nucleotide of exon 26 (3538G>A) causes skipping of 2 non adjecent exons. Blood. 2004; 104: 2739-2745.
- Budde U, Schneppenheim R. Eikenboom J, Goodeve A, Will K, Drewke E, et al. Detailed von Willebrand factor multimer analysis in patients with von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of type 1 von Willebrand disease (MCMDM- 1VWD). J Thromb Haemostas. 2008; 6: 762-771.
- Schneppenheim R, Markgraf O, Eckert K, Obser T, Oyen F, Pieconka A, et al. Molecular background of ‚smeary’ von Willebrand factor multimers. Annual Meeting American Society of Hematology. Blood. 2007; 110: 709-2711.
- Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of type 1 von Willebrand Disease (MCMDM-1VWD). Blood. 2007; 109: 112-121.
- Castaman G, Lethagen S, Federici AB, Tosetto A, Goodeve A, Budde U, et al. Response to desmopressin is influenced by the genotyp and phenotype in type 1 von Willebrand disease (VWD): results from the European study MCMDM-1VWD. Blood. 2008; 111: 3531-3539.
- Schneppenheim R, Lenk H, Obser T, Oldenburg J, Oyen F, Schneppenheim S, et al. Recombinant expression of mutations causing von Willebrand disease type Normandy: characterization of a combined defect of factor VIII binding defect and multimerization. Thromb Haemostas. 2004; 92: 36-41.
- Casonato A, Sartorello F, Cattini MG, Pontera E, Soldera C, Bertomoro A, et al. An Arg760Cys mutation in the consensus sequence of the von Willebrand factor propeptide cleavage site is responsible for a new von Willebrand disease variant. Blood. 2003; 101: 151-156.
- Hilbert L, Nurden P, Caron C, Nurden AT, Goudemand J, Myer D, et al. Type 2N von Willebrand disease due to compound heterozygosity for R854Q and a novel R763G mutation at the cleavage site of von Willebrand factor propeptide. Thromb Haemostas. 2006; 96: 290-294.
- Mannucci PM. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first twenty years. Haemophilia. 2000; 6: 60-67.
- Mannucci PM, Aberg M, Nilsson IM, Robertson B. Mechanism of plasminogen activator and factor VIII increase after vasoactive drugs. Br J Haematol. 1975; 30: 81-93.
- Holmberg L, Nilsson IM, Borge L, Gunnarsson M, Sjorin E. Platelet aggregation induced by 1-desamino-8-D-arginine vasopressin (DDAVP) in Type IIB von Willebrand’s disease. N Engl J Med. 1983; 309: 816-821.
- Gadisseur A, Berneman Z, Schroyens W, Michiels JJ. Laboratory diagnosis of von Willebrand disease type 1/2E (2A subtyoe IIE), typr1 Vicenza and mild type 1 caused by mutations in the D3, D4, B1-B3, and C1-C2 domains of the von Willebrand factor gene. Acta Haematol. 2009; 121: 128-138.
- Federici AB, Mazurier C, Berntorp E, Lee CA, Scharrer I, Goudemand J, et al. Biologic response to desmopressin in patients with severe type 1 and type 2 von Willebrand disease: results of a multicenter European study. Blood. 2004; 103: 2032-2038.
- Lethagen S, Harris AS, Sjorin E, Nilsson IM. Intranasal and intravenous administration of desmopressin: effect on FVIII/VWF, pharmacokinetics and reproducibility. Thromb Haemost. 1987; 58: 1033-1036.
- Kohler M, Hellstern P, Tarrach H, Bambauer R, Wenzel E, Jutzler GA. Subcutaneous injection of desmopressin (DDAVP): evaluation of a new, more concentrated preparation. Haemostasis. 1989; 19: 38-44.
- Michiels JJ, van Vliet HH, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, et al. Intravenous DDAVP and FVIII-von Willebrand factor concentrate for the treatment and prophylaxis of bleeding in von Willebrand disease type 1, 2 and 3. Clin Applied Thromb/Hemostas. 2007; 13: 14-34.
- De La Fuente B, Kaspar CK, Rickless FR, Hoyer LW. Response of patients with mild and moderate hemophilia and von Willebrand disease to treatment with desmospressin. Ann Intern Med. 1985; 103: 6-14.
- Millar CM, Riddell AF, Brown SA, Starke R, Mackie I, Bowen DJ, et al. Survival of von Willebrand factor released following DDAVP in a type 1 von Willebrand disease cohort: influence of glycolysation, proteolysis and gene mutations. Thromb Haemost. 2008; 99: 916-924.
- Huang RH, Wang Y, Roth R, Yu X, Purvis AR, Heuser JE, et al. Assembly of Weibel-Palade body-like tubules from N-terminal domains of von Willebrand factor. Proc Natl Acad Sci USA. 2008; 105: 482-487.
- Haberichter SL, Balistrieri M, Christopherson P, Morateck P, Gavazova S, Bellissimo DB, et al. Assay of von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006; 108: 3344-3351.
- Haberichter SL, Castaman G, Budde U, Peake I, Goodeve A, Rodeghiero F, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor (VWF) survival by assay of the VWF propeptide in the European study: Molecular and Clinical Markers for the Diagnosis and Management of type 1 VWD (MCMDM-1VWD). Blood. 2008; 111: 4979-4985.
- Davies JA, Collins PW, Hathaway LS, Bowen DJ. Von Willebrand factor: evidence for variable clearance in vivo according to Y/C1584 phenotype and ABO blood group. J Thromb Haemost. 2008; 6: 97-103.
- Michiels JJ, Gadisseur A, van der Planken M, Schroyens W, van de Velde A, Berneman Z. Guidelines for the evaluation of intravenous desmopressin and von Willebrand/FactorVIII concentrate in the treatment and prophylaxis of bleedings in von Willebrand disease types 1, 2 and 3. Sem Thromb Hemost. 2006; 32: 636-643.
- Gadisseur A, Michiels JJ. Advances in the diagnosis and classification of von Willebrand disease. Acta Haematol. 2009; 121: 67-186.
- Vangenechten I, Mayger K, Smejkal P, Zapletal D, Michiels JJ, Moore GW, et al. A comparative analysis of different automated von Willebrand factor GPIb-binding activity assays in well typed von Willebrand Disease patients. J Thromb Haemostas. 2018; 16: 1-10.
- Vangenechten I, Smejkal P, Zapletal O, Michiels JJ, Berneman Z, Zavrelova J, et al. Analysis of von Willebrand Disease in the South Moravian population (Czech Republic): Results from the BRNO-VWD Study. Thromb Haemostas. 2019; 119: 594-606.
- Michiels JJ, Smejkal P, Mayger K, Moore G, Budde U, Berneman Z, et al. Combined use of rapid von Willebrand factor (VWF) activity , VWF-propeptide and classical VWF assays for improved diagnosis of von Willebrand disease type 1, 2N, 2 E, due to mutations in the D1, D2, D3 and D4 domains of the VWF gene. Thromb Haemost Res. 2019; 3: 1027
- Michiels JJ, Smejkal P, Mayger K, Moore G, Blatny J, Penka M, et al. Superiority of the rapid Glycoprotein GPIbR and GPIbM von Willebrand factor (VWF) activity assays in well defined von Willebrand disease type 2A, 2B,and 2M due to mutations in the A1, A2 andA3 domain of the VWF gene. Hemostas Thromb Res In press.
Citation: Smejkal P, Jacques Michiels J, Zapletal O, Blatny J, Batorova A, Pricangova T, et al. European Clinical, Laboratory and Molecular (2020 ECLM) Diagnostic Work-Up and Classification of Von Willebrand Disease from the Perspectives of Clinicians and Scientists. Thromb Haemost Res. 2019; 3(3): 1032.