
Research Article
Thromb Haemost Res. 2019; 3(4): 1035.
Reinvestigation of Synthesis of Enoxaparin Under PTC Conditions
Venkatanarayana P1, Prasanna B2* and Nareshvarma Seelam1
¹Department of Chemistry, Koneru Lakshmaiah Education Foundation, India
²Department of Chemistry, Chaitanya Post Graduate College (Autonomous), India
*Corresponding author: Prasanna B, Department of Chemistry, Chaitanya Post Graduate College (Autonomous) Kishanpura, Hanamkonda, Warangal, Telangana State 506 001. India
Received: November 04, 2019; Accepted: December 05, 2019; Published: December 12, 2019
Abstract
Heparin is a mixture of Glycosaminoglycan (GAG) chains originating from porcine intestinal mucosa. It is used therapeutically as an anticoagulant for the treatment and prevention of thrombosis. Glycosaminoglycans such as Heparin (H) and Heparan Sulfate (HS) are considered attractive therapeutic agents because they modulate many biological processes and have been implicated in numerous pathologies, including cardiovascular, cancer, inflammation, metabolic, and neurodegenerative diseases and viral infections. These biological functions are believed to be dependent on the interaction of these linear polysaccharides with key proteins such as growth factors, cytokines, proteases, adhesion proteins, lipid binding proteins, etc., which have a heparin binding domain in common and are termed Heparin Binding Proteins (HBPs). The variability in unfractionated heparin’s pharmacokinetic properties and pharmacological effects led to the development of Low MW Heparin (LMWH), which is a degraded product of heparin using chemical or enzymatic cleavage techniques. The most common form of LMWH in the U.S. is enoxaparin, which is produced by β-eliminative cleavage of the benzyl esters of porcine mucosal heparin under alkaline conditions. This cleavage process leads to the generation of unnatural structures in enoxaparin. We present herein the purification strategies used to generate hexasaccharides that were further evaluated in vitro for their affinity for these protein targets, as well as heparanase inhibition. The hexasaccharides contain the same (L-iduronic acid-D glucosamine) carbohydrate backbone but varying substitution patterns. We present here a new purification process of Enoxaparin with good yield.
Keywords: Heparin; Enoxaparin; Low molecular weight; Anticoagulant
Introduction
The anticoagulant drug Heparin has been used to treat thrombosis for a propos 80 years [1]. The drug was originally isolated from dog liver and demonstrated to possess anticoagulant activity in 1916 [2]. During the 1930s, heparin was successfully prepared from bovine lung, and this drug source was later developed as a pharmaceutical product in the United States [3,4]. Heparin is a linear polysaccharide composed of a repeating disaccharide building block of alternating β-1,4-linked Hexuronic Acid (HexA) and Glucosamine residue (GlcN). The HexA can be either β-D-Glucuronic Acid (GlcA) or a-LIduronic Acid (IdoA) at which the C-2 position can be substituted by an O-sulfo group. The GlcN may be modified by an N-acetyl group (GlcNAc), an N-sulfo group (GlcNS), or can be un-substituted, whereas O-sulfo group substitution can occur at its C-3 and/or C-6 positions [5,6]. A pentasaccharide sequence of GlcNAc/NS(6S)-GlcAGlcNS( 3S,6S)-IdoA(2S)-GlcNS(6S) is the structural motif for heparin that specifically binds to antithrombin III (ATIII) and inactivates the blood clotting process [7].
Heparin molecule comes under the family of Glycosaminoglycans (GAGs) and consists of the heterogeneous mixture of polymer due to this the molecule has been referred to as “Unfractionated Heparin (UFH) amongst clinicians and researchers [8]. Heparin possesses biological functionality towards angiogenesis and hostpathogen interactions [9-11]. Furthermore, Heparin is popular in the pharmaceutical industry for its anticoagulant properties, and its de-polymerized version, termed Low Molecular Weight Heparins (LMWH), has gained much attention in the recent past. Different types of LMWH are derived based on differing de-polymerization methods. These molecules are similar to UFH in monosaccharide composition and oligosaccharide sequence. LMWH possess several advantages over UFH due to their lower molecular mass, including prolonged antithrombotic effect and better bioavailability. Given that LMWH do not bind to plasma proteins and endothelial cells, they have a longer half-life in circulation [12]. Due to the often-reported side effects causing Heparin Induced Thrombocytopenia (HIT), LMWHs have been explored as anticoagulants13. These molecules are considered more potent compared to Unfractionated Heparin (UFH) [14].
In this paper, we describe the significant procedure for the synthesis of the Enoxaparin, leading to a simplification and shortening of the process. The synthesis steps have been shown in Scheme-1. We have used a method for the de-polymerization of Heparin sodium to get Enoxaparin, and have characterized the synthetic intermediates in their production. It is our hope that the data presented here and the convenience of our method will facilitate further investigation of this important class of compounds. The spectral and analytical data strongly supported the structure of heparin.
Results and Discussion
Routes used to de-polymerization of the compound described in this work, are depicted in Scheme 1. Key intermediates 2, 3 were prepared from corresponding Heparin sodium 1 by the complexation, separation, reduction.
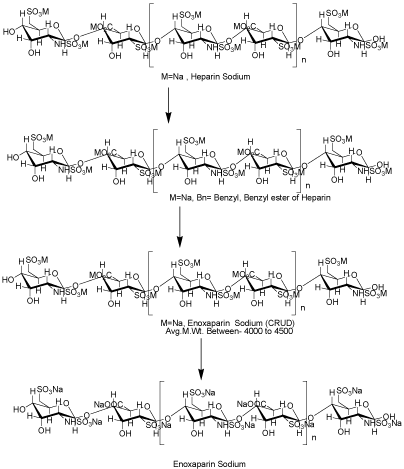
Figure 1: Key intermediates 2,3 were prepared from corresponding Heparin sodium 1 by the complexation, separation, reduction.
Experimental section
Melting points were uncorrected. Infrared spectra were obtained by using a BrukerWM-4(X) spectrometer 577 model [1]. H NMR (400MHz) and [13] C NMR (100MHz) spectra were recorded on a Bruker WM-400 spectrophotometer in CDCl3 with tetramethylsilane as reference. Mass spectra (ESI) were carried out on a JEOL SX-102 spectrophotometer. Elemental analysis was done on a Carlo Erba EA 1108 automatic elemental analyzer. The chemicals and solvents were commercial grade, used without further purification. Purification of the synthesized compounds by column chromatography and Thin- Layer Chromatography (TLC) was carried out by using alumina sheets purchased from Merck with ethanol as the moving phase.
General procedure for the synthesis of Enoxaparin stages
Stage 1: Taken heparin sodium (1.0mol) was completely dissolved in purified water (100mL) at 30-35oC. Check the PH of reaction mass, should be 6.8 to 7.2 ranges. If the PH range is increases adjusted by using 5N NaOH solution and Sodium Sulphite was charged to heparin sodium solution. The reaction mass stirred for 10 minutes at 30-35oC. Took another round bottomed flask catalyst Cetrimide (PTC) was completely dissolved in purified water at 30-35oC. The prepared heparin solution was added to cetrimide solution and followed by stirring at 30-35oC for 0.5h, after maintained the reaction mass settled for 3.0h solid separated was filtered washed with purified water and MDC for two times to get the corresponding compound.
Stage 2: To the round bottomed vessel added Stage-1 (0.1mmol) material dissolved in MDC (100mL) at 35-40oC in 1.5h, cool to reaction mass than a solution of benzyl chloride (0.12mmol) was added. The reaction mass stirred for 18.0 to 40.0 h at 35oC. The reaction mass was cooled to room temperature than added MDC and Methanol was added. Sodium acetate was added to reaction mass, followed by water maintained for 0.5h. Gummy material was formed decant the organic layer washed with methanol (5x20mL). The solid material dried under vacuum dried at 50-550C.
Stage 3: To a stirred solution of Stage-2 (0.1 mmol) compound in purified water added anhydrous Sodium Sulphite and EDTA salt was charged. The reaction mass was stirred for 15 min, heated to 60-65oC. Added 5N sodium hydroxide solution maintained for 0.5h. The reaction mixture was cooled to 30-40oC than adjusted PH neutral by 6N aq. HCl. Ethanol was added to reaction mass and stirrer for 15 minutes settled the reaction mass for 1h, gummy material was formed. The solvent layer was separated and dehydrates with methanol the solid material was filtered and dried under vacuum at 600C.
Stage 4: Stage-3 (0.1mmol) compound dissolves in purified water (100mL) adjusted PH by using 5N NaOH solution to 8.0-9.0 range. Heat the reaction mass up to 50oC added hydrogen peroxide stirred for 1h. The reaction mass was cooled to 30-35oC, check the PH of reaction mass and adjusted by using 5N NaOH solution (PH 6.0-6.5). Stirred for 10 min and filtered through hyflo bed under Nitrogen gas EtOH was added to reaction mass and stirred for 15 min. Separate the solvent layer from reaction mass and added MeOH to gummy material to get the purified compound.
Characterization of enoxaparin
Off white crystalline powder, U.V. 232 nm; 1H NMR (600MHz): 0.10-2.00; 2.10-3.20; 5.70-8.00; 3.35-4.55; 13C NMR(300MHz): 176.47,176.09; 175.66;170.42, 145.97, 107.099, 103.01, 100.27,98.57, 98.03, 94.05, 92.36, 79.39,78.45, 77.01, 76.49, 75.81, 75.44, 74.71, 74.05,72.24, 71.88, 70.80, 70.35, 69.99, 68.49, 67.59, 64.20, 63.91, 61.05, 59.17, 58.83, 50.31, 49.78, 49.49, 49.20, 48.92, 48.63, 23.29, 18.18. Average molecular weight: 4345 (Range: 3800-5000Da) ‹2000Da:14.5% (Range: 2.0%-20.0%), 2000-8000Da:72.0% (Range:-68.0%-82.0%) >8000 Da: 8.61% (Range:-NMT 18.0%). Antifactor Xa (on dry basis):108 IU/mg (Range: 90-125 IU/mg). Antifactor IIa (on dry basis): 28 IU/mg (Range: 20-35 IU/mg), The ratio of anti-factor Xa / IIa: 3.857 (Range: 3.3-5.3) Molar ratio of sulphate ions to carboxylate ions: 2.2 (less than 1.8), PH -10% solution in water: 6.6(Range: 6.2-7.7). Specific absorbance at 231nm:15.8% (Range: 14.0-20.0). Loss on drying: 2.48 % (not more than 10.0%). Heavy metals (not more than 30ppm): Compiles.
References
- Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002; 41: 390-412.
- Mclean J. The thromboplastic action of cephalin. Am J Phys. 1916; 41: 250- 257.
- Charles AF, Scott DA. Studies on heparin I. The preparation of heparin. J Biol Chem. 1933; 102: 425-429.
- Charles AF, Scott DA. Studies on heparin II. Heparin in various tissues. J Biol Chem. 1933; 102: 431-435.
- Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat. Prod. Rep. 2002; 19: 312-331.
- Casu B. Structure of heparin and heparin fragments. Ann N Y Acad Sci.1989; 556: 1-17.
- Lindahl U, Thunberg L, Backstrom G, Riesenfeld J, Nordling K, Bjork I. Extension and structural variability of the antithrombin-binding sequence in heparin. J. Biol. Chem. 1984; 259: 12368-12376.
- Guerrini M, Bisio A. Low-molecular-weight heparins: differential characterization/ physical characterization, Hand b Exp. Pharmacol. 2012; 127-157.
- Alam F, Hwang SR, Al-Hilal TA. Safety studies on intravenous infusion of a potent angiogenesis inhibitor: taurocholate-conjugated low molecular weight heparin derivative LHT7 in preclinical models. Drug Dev Ind Pharm. 2016; 42: 1247-1257.
- Thacker BE, Seamen E, Lawrence R, Parker MW, Xu Y, Liu J, et al. Expanding the 3-O-Sulfate Proteome-Enhanced Binding of Neuropilin-1 to 3-O-Sulfated Heparan Sulfate Modulates Its Activity. ACS Chem Biol. 2016; 11: 971-980.
- Xu Y, Martinez P, Seron K, Luo G, Allain F, Dubuisson J, et al. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J Virol. 2015; 89: 3846-3858.
- Aguilar OM, Kleiman NS. Low molecular weight heparins. Expert Opin Pharmacother. 2000; 1: 1091-1103.
- Chandarajoti K, Liu J, Pawlinski R. The design and synthesis of new synthetic low molecular weight heparins. J Thromb Haemost. 2016; 14: 1135-1145.
- Casu B, Naggi A, Torri G. Re-visiting the structure of heparin. Carbohydr Res. 2015; 403: 60-68.