
Review Article
Thromb Haemost Res. 2020; 4(3): 1049.
Comparative Analysis of the European, Canadian and UK ISTH Defined VWD Type 1 Prospective Studies: Principles to Translate the ISTH into the ECLM Classification of Von Willebrand Disease on Top of Sensitive Von Willebrand Factor Assays and Multimeric Analysis in Medium to High SDS Resolution Gels
Michiels JJ1,2,3*, Smejkal P1, Penka M1, Budde U4, Hermans C5, Blatny J6, Batorova A7, Pricangova T7, Gadisseur A8, Vangenegten I8, Moore G9, Mayger K9 and Van Vliet H2,3
1Department of Clinical Haematology, Masaryk University, Czech Republic
2Department of Hematology Hemostasis and Thrombosis Research, Erasmus University Medical Center Rotterdam, The Netherlands
3Goodheart Institute in nature Medicine & Health, Blood Coagulation and Vascular Medicine Center, The Netherlands
4Central Laboratory, Asklepios Kliniken, Germany
5Hemostasis Thrombosis Unit Haemophilia Clinic, St-Luc University Hospital Belgium
6Department of Pediatric Haematology, Children’s University Hospital, Czech Republic
7Department of Hematology, University Hospital and Medical School of Comenius University, Slovakia
8Department of Hematology and Hemostasis Research Unit, Antwerp University Hospital, Belgium
9Department of Haemostasis Thrombosis, Viapath Analytics at Guys and St Thomas NHS Foundation Trust, UK
*Corresponding author: Jan Jacques Michiels, Associate Professor Huub Van Vliet, Goodheart Institute in nature Medicine & Health, Blood Coagulation and Vascular Medicine Center, Freedom of Science and Education, Erasmus Tower, Veenmos 13, 3069 AT Rotterdam, The Netherlands
Received: May 12, 2020; Accepted: June 18, 2020; Published: June 25, 2020
Abstract
A complete set of Von Willebrand Factor (VWF) laboratory investigations including bleeding time, PFA-100 closure times, FVIII: C, VWF Ristocetin Cofactor activity (VWF: RCo). VWF Collagen Binding (VWF: CB), VWF Antigen (VWF: Ag), Ristocetin Induced Platelet Aggregation (RIPA), VWF multimeric analysis and the response of FVIII: C and VWF parameters to DDAVP have the power to diagnose all variants of Von Willebrand Disease (VWD) and to discriminate between the type 1 and type 2 and between severe type 1 and type 3 VWD. The response to DDAVP of VWF parameters is normal in pseudo-VWD (mild VWF deficiency due to blood group O), in mild VWD type 1 and in carriers of type 1 and 3 VWD. The response to DDAVP is rather good but restricted followed by increased clearance in dominant type 1/2E, transiently good in mild type 2A group II, good for VWF: CB but poor for VWF: RCo in 2M and 2U, poor in 2A group I, 2B, 2C and 2D, and very poor or non-responsive in recessive VWD severe type 1 and 3.
Homozygosity or double heterozygosity for non-sense mutations in the VWF gene are the cause of recessive VWD type 3. Homozygosity or double heterozygosity for a non-sense and mis-sense mutation or for two missense mutation cause recessive severe type 1 VWD. VWD type 3 is a hemophilialike bleeding disorder with the complete absence of VWF and FVIII: C and compatible with life. Recessive “type 1” VWD differs from “type 3” VWD by the presence of detectable VWF: Ag and FVIII: C levels between 0.01 and 0.10 U/dL. Heterozygous carriers of recessive type 3 or 1 VWD with one mutant null or missense allele are usually asymptomatic at VWF levels around 50% of normal. Recessive type 2C (IIC) is due to homozygous or double heterozygous mutations in the D2 domain. Homozygosity or double heterozygosity for the FVIII binding defect of the VWF is the cause of recessive VWD type 2 Normandy featured by low FVIII: C, mild or moderate VWF deficiency type 1 VWD and normal VWF multimers. Heterozygous carriers of VWD type 2C (IIC) may have mild VWF: RCo deficiency, mild bleeding and abnormal 2C like multimer defect in high resolution gel.
LowVWF mild VWD type 1 patients present with low to variable penetrance of bleeding, have prolonged PFA-CT between the upper limiy of normal to 300 seconds, show a high (increased) prevalence of blood group O, have VWF values between 0.30 and 0.60 U/dL with normal ratios of VWF: RCo/: Ag, VWF: CB/Ag and FVIII: C/VWF: Ag. LowVWF mild VWD type 1 has low penetrance of mucocutaneous bleeding, the combination of C1584/blood group O is rather frequent in the Euopean and Canadian VWD-1 studies. LowVWF mild VWD type 1 show good and adequate responses of FVIII: C and VWF parameters to DDAVP. The presence of a missense mutation in heterozygous LowVWF mild type 1 VWD patients are featured by normal multimers in a low resolution gel and are mainly located in the regulatory sequence region, the D1 D2 region, the D’ VWF-FVIII binding site region and the D4, C1 to C6 region of the VWF protein, but less frequent to rarely in the D3, A1 or A2 domain. A new category of mild VWD type 1 due to mutations in the D4, B1-3, C1-2 (recently labelled as C1 to C6) domains of the VWF gene has been discovered by the European MCMDM-1VWD study and consist of two groups. One group have normal VWF multimers and present with mild VWD either dominant or recessive type 1 VWD with variable penetrance of bleeding manifestations. The other group of VWD type 1 with mutations in the D4, B1-3, C1-2 (C1 to C6) domains have a smeary pattern of abnormal VWF multimers in low and medium SDS resolution gels.
Autosomal dominant VWD type 1/2E (IIE) is quantitative/qualitative multimerization defect caused by a heterozygous cysteine mutation in D3 domain of the VWF gene resulting in a secretion (increased FVIII: C/VWF: Ag ratio) and/or clearance defect (increased VWFpp/Ag ratio). The VWF in VWD 1E/2E VWD patients lack the triplet structure on VWF multimers using medium to high resolution gel according to the method of Budde. Dominant VWD type 1/ Vicenza is qualitative defect with equally low levels of FVIII: C, VWF: Ag, VWF: RCo, VWF: CB due to rapid clearance (high VWFpp/Ag ratio) with the presence of large VWF multimers in plasma caused by a specific mutation R1205H in the D3 domain. VWD 2A variant IIA, IIB, IIC, and IID are featured loss of large VWF multimers in low resolution gels and caused by increased proteolysis in VWD type 2A (IIA) and 2B (IIB) RIPA is decreased in 2A (IIA) and increased in IIB. VWD 2B (IIB) is caused by a gain of GPIb function mutation and VWD 2D (IID) is a dimerization defect due to mutation in the CK domain. Dominant VWD type 2M and 2U (2A-variant) are caused by loss of function mutations in the A1 domain resulting in a quantitative/qualitative variants featured by decreased RIPA, decreased platelet dependent function VWF: RCo, and normal VWF: CB with the presence all VWF multimers (VWD 2M). VWD 2U (2A-variant) VWD patients have some loss or relative decrease of large VWF multimers (VWD 2A variant). Proteolysis of VWF is minimal in each of type 2C (IIC), 2D (IID), 2E (IIE), and 2M or 2U. VWD 2M and 2A-variant have aberrant VWF multimeric structure of individual oligomers and lack of triplet structure of each band. Proteolysis of VWF is increased in dominant type 2A and 2B VWD and result in the absence of large VWF multimers, increased triplet structure of each band, decreased ratios for VWF: RCo/Ag and VWF: CB/Ag and prolonged BT. Autosomal dominant type 2A (IIA) is caused by heterozygous missense mutations in the A2 domain. VWD type 2B (IIB) is due to gain of function mutations in the A1 domain and differs from 2A by a normal VWF multimeric pattern in platelets, and increased RIPA.
Introduction
In routine daily practice simple sensitive and specific Von Willebrand Factor (VWF assays) are to be used for clinical suspicion and grading of bleeding severity evaluation and correct diagnosis of patients with Congenital Von Willebrand Disease (VWD) [1- 12]. In classic VWD type 1, 2 or 3, either autosomal recessive or dominant, the patient has recurrent mucocutaneous since early childhood, more than two bleedings after tooth extraction, trauma or surgery and bleedings that needed medical treatment and/or FVIII/VWF concentrate transfusion because of abnormal bleeding after an operation and/or trauma, menarche (in women), or has bleed for a few to several hours or even more than 24 hours after a tooth extraction, minor trauma or surgery. A moderate to severe type of mucocutaneous bleeding either recessive or dominant since early childhood plus, hemarthrosis, muscle bleeding, and a need for prophylactic treatment with FVIII/VWF concentrate, refers to a hemophilia bleeding type, which is usually seen in recessive 3 VWD type 3 [1,2]. In very mild von Willebrand factor deficiency related to blood group O and/or VWD type 1 in the study of Michiels et al. (2002) [12] have VWF values between 0.30 U/dL and 0.60 U/ dl (LowVWF mild type 1 VWD), the patient has only one or two unclear minor bleeding symptoms, no serious bleeding in childhood and absence of secondary bleedings following trauma and/or surgery. In LowVWF mild type 1 VWD, the patient has one or two obvious mucocutaneous symptoms like frequent episodes of epistaxis, and/or prolonged or profuse menstruation or frequent hematomas, which usually do not require medical treatment or FVIII/VWF concentrate treatment and usually show a rather good but restricted response to DDAVP in carriers of recessive type 1 and type 3 to normal response of FVIII: C and VWF parameters to DDAVP in blood group O individuals (pseudo-VWD (Michiels et al 2002) [12].
Diagnostic differentiation of VWD type 1 and 2 with “normal” VWF multimers
The ISTH classification of congenital Von Willebrand Disease (VWD) is dominated by the recommendations of the VWF Scientific Standardization Committee (VWF-SSC) at annual SSC meetings of the International Society on Thrombosis and Haemostasis (ISTH) between 1994 and 2000 [10-12]. The 1994-2000 ISTH classification is based on a small set of insensitive VWF parameters VWF: Ag, VWF: RCo, RIPA and VWF multimers in a low resolution gel. VWF multimeric analysis in low SDS resolution gels cannot distinguish the 2A (IIA, IIB, IIC, IIE and IID variants. VWF multimeric analysis in medium to high resolution gels used by Ruggeri & Zimmermann, by Battle et al and by Budde & Schneppenheim is highly sensitive for the diagnosis of VWD type 1 with normal VWF multimers and does distinguish each of VWD subtypes type IIA, IIB, IIC, IID and 2M related to mutations in the A2, A1, D2, D3 and CK domain respectively (Figure 1) [1-12]. The VWF-SSC classification of VWD patients is based on a few insensitive laboratory tests including FVIII: C, VWF: Ag, VWF: RCo, VWF: RCo/Ag ratio RIPA and VWF multimers in low resolution gels and therefore persisted to lump VWD 2A variants IIA, IIB, IIC, IIE and IID as one category (Table 1). The lowest detection level of the VWF;RCo assay between 1994 and 2000 was 0.10 to 0.15 U/dl), and RIPA was only used for the differentiation of VWD IIA (2A) and IIB (2B), whereas VWF multimers in low resolution gel only detect the absence of large VWF multimers. For reasons of applicability in routine practice, the SSC-ISTH used the combined “lumping” rather than the “splitting” approach for the classification of type 2 VWD based on arbitrary rules but not based on scientific analysis of structure and function relationship of VWF gene mutations related to the functional domains of the VWF protein.
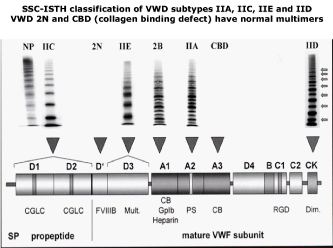
Figure 1: 2000 - 2006 SSC-ISTH Classification of VWD type 2A and its subtypes IIA, IIC, IIE and IID [11,13].
![]()
- Inherited VWD caused by genetic mutations at the VWF locus includes a broad spectrum of recessive and dominant variants of VWD. Carriers of recessive VWD type 3 or severe recessive type 1 VWD are asymptomatic or may manifest mild bleeding in particular when associated with blood group O1,2,.
- Type 1 VWD refers to partial quantitative deficiency of VWF mainly based on a normal VWF:RCo/Ag ratio VWD type 2A (lumping IIA, IIB, IIC, IIE,IID) VWD refers to qualitative VWF deficiency with the loss of large VWF multimers as documented by a decreased VWF:RCo/Ag ratio. VWD type 3 refers to virtually complete deficiency of VWF and FVIII:C.
- Type 2A VWD refers to qualitative variants with decreased platelet dependent function that is attributed to the absence of high molecular weight VWF multimers. The 2006 SSC ISTH followed a compromised “lumping-spitting” approach in distinguishing four mains categories of type 2A (IIA), 2B (IIB), 2M, 2N VWD patients (Figure 1).
- Type 2M or 2U is a new entity defined by the 1994-2000 SSC-ISTH and refers to qualitative variants with decreased platelet dependent function (VWF:RCo and RIPA), with the presence of large VWF multimers in a low resolution agarose gel (VWD 2M), but recent data show a relative decrease of large VWF multimers in a medium resolution gel (VWD 2A-variant).
- Type 2A VWD refers to qualitative variants with absence of HMW multimers, increased, normal or decreased RIPA and decreased VWF:RCo/Ag ratio. The lumping-splitting approach 2A by the ISTH 2006 can be subdivided in the variants of IIA, IIC, IIE and IID as defined by Zimmerman & Ruggeri, Battle, Budde & Schneppenheim (Figure 1).
- Type 2B (IIB) refers to all qualitative variants with absence of HMW multimers and decreased VWF:RCo/Ag ratio and increased RIPA as first described by Ruggeri & Zimmerman.
- Type 2N (Normandy) refers to all qualitative variants with markedly decreased binding of factor VIII to VWF. VWD 2N show the presence of all large and intermediate VWF multimers with a normal VWF:RCo/Ag ratio as first discovered as VWD Normandy (2N)l [17-20].
Table 1: Classification of VWD according to the VWF-SSC-ISTH guidelines 1994-2002 [10,11,13].
In the “lumping” approach of the ISTH classification, VWD type 2 is independent of multimer structure in a low resolution gel and refers to all loss-of-function variants of VWF including FVIII binding defect (FVIII: B defect = Normandy = 2N), loss of high molecular weight (HMW) multimers with decreased RIPA in VWD 2A lumped as VWD IIA, IIC, IID, or loss of HMW and increased RIPA VWD 2 (IIB). Lumping is easier to apply at the expense of illogical grouping of several variants of VWD IIA, IIC, IID, IIE as 2A with decreased RIPA and loss of large VWF multimers due to various mechanisms underlying VWD type 2. The underlying mechanisms of VWD type 2 include increased proteolysis in VWD type 2A (IIA) and 2B (IIB) due to mutations in the A2 and A1 domain respectively; multimerization defect in VWD 1E/2E (IIE) due to mutations in the D3 domain, and dimerization defect in VWD 2D (IID) due to mutations in the CK domain. The ISTH 1994 and 2000 ISTH classification poorly defined dominant VWD 2M and its differentiation from dominant pronounced type 1 was hardly possible VWF levels below 0.20 U/ dL. In 2000 decreased or absent RIPA due to loss of function in the interaction between platelet-GPIb-ligand and VWF-GPIb receptor appeared to key feature of VWD 2M and 2U with normal but relative loss of large multimers, whereas RIPA is normal in pronounced dominant VWD type 1. Initially VWD type Vicenza has labeled as 2M but the detection all VWF multimers and normal RIPA after DDAVP followed clearance VWF: Ag and FVIII: C changed te classification of VWD Vincenza into VWD 1C. Recessive VWD 2N (Normandy) is caused by a FVIII-VWF binding defect but has normal VWF multimers typical for VWD type 1.
In the “spitting” alternative, type 2 VWD refers to the loss-offunction (VWF: RCo) of circulating VWF attributed to the absence of HMW multimers in 2A with decreased RIPA (lumping IIA, IIC, IIE and IID with decreased RIPA) and 2B (IIB) with increased RIPA according to Ruggeri & Zimmerman [1,2]. VWD type 2M frequently labeled as 2U or 2A-variant refers to loss of VWF GPIb function (decreased VWF: RCo and decreased RIPA) not caused by the loss of HMW multimers. Using the 1994 – 2000 SSC-ISTH criteria, the diagnosis of pronounced dominant VWD type with VWF: Ag and VWF: RCo levels below 0.15 U/L could not be differentiated from dominant VWD type 2 M and also not from recessive pronounced VWD type 1 in the study of Michiels & Van Vliet 2002 [12-14]. The original description of dominant VWD type 2E (IIE) usually has a laboratory phenotype 1 in all VWD studies when the ISTH classification is applied [10-62]. Diagnostic differentiation of so-called pronounced dominant or recessive VWD type 1 using the 1994-2006 SSC-ISTH criteria remained a persistent problem in routine daily practice [10-62].
Fressinaud et al. (1998) evaluated the performance of the PFA- 100 Closure Times (CT) in 60 patients with VWD classified according to the 1994 SSC-ISTH classification15. This study included 36 patients with VWD type1, 24 patients with VWD 2A (IIA), 2B (IIB), patients with 2N, and in 14 patients with hemophilia (Table 2). PFA CT Epi and CT ADP were normal in VWD 2N and in hemophilia patients. According to strict criteria proposed by Michiels et al the VWD type 1 patients could be reclassified as mild VWD type 1 in 15 cases (VWF: RCo 0.28-0.39 U/dl), moderate VWD type 1 or 2 in 9 cases (VWF: RCo 0.15-0.27 U/dl) and severe VWD type 1 or 2 in 12 cases (VWF: RCo 0.05-0.14 U/dl). All patients with severe VWD type 1 or 2, 2A (IIA), 2B (IIB) and 3 had prolonged PFA CT unmeasurable above 250 seconds and prolonged cutaneous BT in the majority. The Bleeding Time (BT) varied from normal in a few to prolonged in patients with moderate or severe type 1 or 2 VWD. Mild VWD type 1 patients had normal to marginally prolonged BT and slightly prolonged PFA -CT between the upper limit of normal and 250 seconds. These observations lead to the conclusion that the PFA-100 is clearly superior to cutaneous BT for the detection of mild versus pronounced VWD. A normal PFA-CT exclude VWD except VWD type 2N.
![]()
Patients
VWF:
Ratio
VWD type
BT
PFA-100 CT seconds
VWD type
Ag
Rco
Rco/Ag
reclassified
ADP
Epi
SSC-ISTH
2002 Michiels
Type 1 mild LowVWF
1
48
39
0.81
1 mild
10
136
137
2
40
39
0.98
1 mild
8
147
173
3
34
38
1.12
1 mild
6
154
202
4
62
38
0.61
1 mild
8.5
148
164
5
39
36
0.92
1 mild
18
193
159
6
45
35
0.77
1 mild
6
141
184
7
37
34
0.92
1 mild
6
144
183
8
35
34
0.97
1 mild
16
210
>250
9
38
34
0.89
1 mild
5
145
218
10
45
34
0.76
1 mild
6
138
180
11
42
31
0.74
1 mild
5.5
127
174
12
38
30
0.79
1 mild
8
129
191
13
35
28
0.8
1 mild
18
197
>250
14
29
28
0.97
1 mild
6
170
>250
15
48
27
0.56
1 mild
3.5
141
219
Type 1 or 2 moderate
16
59
27
0.56
1 or 2
>20
>250
>250
17
31
26
0.84
1 moderate
7
>250
>250
18
38
26
0.68
1 moderate
17
>250
>250
19
28
22
0.79
1 moderate
15
>250
>250
20
23
22
0.96
1 moderate
12
>250
>250
21
39
22
0.56
2
17
181
>250
22
30
22
0.73
1 moderate
15
181
>250
23
38
16
0.42
2
8.5
197
>250
24
24
15
0.62
1 moderate
6.5
>250
>250
Type 1 or 2 severe
25
24
14
0.58
1 or 2 severe
>20
>250
>250
26
44
13
0.33
2 severe
>20
>250
>250
27
26
12
0.46
2 severe
>20
>250
>250
28
44
13
0.3
2 severe
>20
>250
>250
29
23
10
0.43
2 severe
12
>250
>250
30
12
10
0.83
1 severe
9
>250
>250
31
15
10
0.67
1 severe
>20
>250
>250
32
27
10
0.37
2 severe
11
>250
>250
33
12
9
0.75
1 severe
4
>250
>250
34
29
9
0.31
2 severe
8.5
>250
>250
35
9
0
1
1 severe
>20
>250
>250
36
9
5
0.55
1 or 2 severe
7.5
>250
>250
Type 2A
3 patients
75-92
23-43
0.27-0.57
2A mild
8.5-18
>250
>250
7 patients
30-74
<3-43
0.07-0.57
2A
>20
>250
>250
Type 2B
2 patients
51-61
15-17
0.25-0.33
2B
14->20
>25-
>250
Type 3
4 patients
<1
<3
-
3
>20
>250
>250
Type 2N
2 patients
67-83
54-78
0.81-0.94
FVIII:BD
4-5.5
81-94
<150
Hemophilia A
FVIII:C
57-200
54-164
0.80-1.00
12 pts
<1-27
-
63-119
87-138
Hemophilia B
FIX
2 patients
1.5-3
74-190
58-186
0.78-0.89
-
60-109
<130
Normal values
56-207
58-209
>0.60
<8.5
<120
<160
Table 2: Laboratory parameters in patients with hemophilia, von Willebrand disease type 2A, 2B, 2N, type 3 and VWD type 1 classified according to the 1994 SSCISTH classification of VWD in the French study of Fressinaud et al. [17] and reclassified more strictly according to 2000 ISTH recommendations is hardly possible. Analysed by Michiels et al. in 2002.
Fressinaud et al. (1999) evaluated the PFA-100 in the management of 23 mild to moderate VWD patients with DDAVP [16]. Before DDAVP infusion, the level of VWF: Ag ranged from 0.15 to 0.56 (mean 0.33 ) U/dl and that of VWF: RCo from 0.14 to 0.49 (0.29) IU/dl. All VWD patients had prolonged PAF-100 CT Epi vs ADP above 250 seconds in 13 cases and 8 patients had PFA-CT values between the upper limit of normal to 250 s before DDAVP, The PFA-CT corrected to normal one hour after DDAVP in all 8 and 25 VWD patients (Figure 2). There was no further follow-up except in 3 cases showing only transient correction of CT for only a few hours (Figure 2). Such transient corrections of PFA-100 are typically seen in patients with mild to moderate VWD type 1/2E, mild to moderate 2M, 2M-Vicenza, mild 2A (IIA) and in variants of dominant VWD type due to a secretion and/or rapid clearance defect in the 2002 study of Michiels & Van Vliet [12,14].
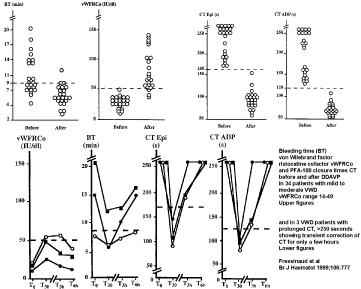
Figure 2: Results of cutaneous Bleeding Time (BT), VWF: RCo and Platelet Function Analyzer Closure Times: PFA CT Epinephrin and PFA CT ADP before and
one hour after intravenous DDAVP in 34 patients with mild to moderate VWD in the French studies of Fressinaud15,16. Please not that half of the 34 VWD patients
did have a prolonged cutaneous BT, whereas PFA CT values were prolonged in all 34 VWD patients and corrected to normal one hour after DDAVP. The figure
below shows that the correction of PFA-CT in VWD patients is transient and lasted only for a few hours.
Michiels reclassified according to ISTH criteria the group of 15 patients with VWD type 1 described by Mannucci et al. (Table 3) [13]. According to improved criteria proposed by Michiels & Van Vliet in 2002 [12], there are 3 cases with severe type 1 VWD patients in Table 1 with low values for FVIII: C and VWF parameters and a very poor response of VWF: RCo and a partial good response of FVIII: C to DDAVP. This type of response as has been described in recessive severe type 1 VWD patients. At least 5 patients in table 1 can readily be reclassified as VWD type 2M based on decreased VWF: Ag and very low VWF: RCo with RCo/Ag ratios of less than 50 when the 2002-2006 Antwerp [1,2,14] criteria and the 2006 SSC-ISTH classification are applied [13]. The response to DDAVP in these type 2M patients is restrictive (not reaching the lower limit of normal, less than 0.60 U/dl) for VWF: RCo, whereas the response of FVIII: C is reasonable normal reaching 2 or 3 times higher to values between 1.0 to 1.5 U/dl. As the ratios of post—to pre DDAVP induced increase of FVIII: C, VWF: Ag and VWF: RCo were not significantly different, this simple means that the decreased ratios of VWF: RCo/Ag ratios in type 2M VWD patients before DDAVP did not correct after DDAVP thereby confirming the diagnosis VWD type 2M [1,2,12,14].
![]()
Patient
Age/Sex
FVIII:CVWF:
VWF:
RCo/Ag
Simplate BT
VWD type
(U/dl)
Ag
RCo
ratio
minutes (<7)
Reclassified
1
36/M
18
12
<6
0.5
15
severe 1 recessive
2
36/F
22
11
9
0.81
7
1
3
29/F
30
58
29
0.5
5
mild 1 or 2
4
55/M
31
8
<6
<0.75
15
severe 1 (2E)
5
53/M
3
20
<6
<0.30
8
severe 1
6
47/M
33
18
7
0.39
12
2M
7
32/F
36
27
<6
0.22
8
2M
8
32/F
40
15
<6
<0.40
20
2M
9
24/F
42
23
23
1
14
1
10
30/M
42
16
6
0.38
11
2M
11
42/F
42
24
29
1.26
5
1
12
19/M
50
15
25
1.66
7
1
13
55/F
55
33
16
0.48
5
mild 1 or 2
14
63/M
57
24
7
0.29
6
2M
15
48/M
12
12
9
0.75
5
severe 1
Table 3: Baseline laboratory data in 15 patients with von Willebrand disease diagnosed as type 1 according to Mannucci et al in 1985 and reclassified according to Michiels et al. [12,14] following the updated 2006 SSC-ISTH criteria [13].
From the French Fressinaud studies in the end 1990s Michiels & Van Vliet concluded in 2002 concluded that the combined use of Ivy BT, VWF;Ag, VWF;RCo, RIPA and VWF multimers in a low resolution gel as recommended by the ISTH absolutely could not distinct between the VWD type 1 versus VWD type 2 in patients with pronounced type 2N, 2M and 1E/2E (IIE) VWD at VWF levels below 0.15 U/ml [12]. To overcome this shortcoming of the ISTH classification Van Vliet & Michiels developed and introduced a novel set of sensitive VWF parameters. The Ivy bleeding time was replaced by Platelet Function Analyzer closure times (PFA-CT ADP and Epinephrine). A normal PFA-CT result exclude mild VWD, The PFA-CT is normal in pseudo-VD, slightly prolonged in mild type 1 VWD and usually strongly prolonged (above 250 second or above 300 seconds). Van Vliet developed the VWF collagen binding ELISA assay using Coll type 1 as described in 2002 (Michiels& Van Vliet) [12]. The VWF: CB assay is sensitive for the presence of large VWF MM in VWD type 1 and VWD type 2M. The VWF;CB is more sensitive than the VWF: RCO assay to detect the loss in large VWF MM in VWD type 2A (IIA, IIC, IIE, IID) and 2A-variant. Van Vliet used the Brosstad method for VWF multimeric analysis as a more sensitive in VWF MM assay in low resolution gels to better separate VWD type 1 and 2M from classical VWD 2A and 2B [12]. Please note that recessive VWD IIC and IID are rare and never seen by Michiels and Van Vliet in the large Rotterdam cohort of VWD patients 1994- 2020 [12]. Michiels & Van Vliet prospectively evaluated between 1994 and 1998 the large Rotterdam cohort of VWD patients study12 the novel set of VWF parameters PFA-CT, FVIII: C, VWF: Ag, VWF: RCo, RIPA, VWF: CB, modified Brosstad VWF analysis on top of a DDAVP challenge test diagnosed as VWD type 1 or VWD type 2 RIPA and a correct interpretation of the response curves of FVIII: C and VWF parameter in subsequent VWD studies [1,2,13,14]. (reviewed in Thrombosis Hemostasis Research 2020: 4(1): 1040).
The molecular basis of mild VWD type 1 with normal VWF multimers: LowVWF type 1 VWD
Parents of patients with recessive VWD type 3 and severe type 1 are carriers of nonsense (null) or missense mutation. Such carriers of heterozygous for the VWF null allele or a missense mutation have no history of bleeding or presented with minor bleedings (one or two bleeding symptoms mainly epistaxis, bruises and/or prolonged menstruations with no abnormal bleeding after tooth extraction, trauma or surgery) [1,21-26]. There is a wide range of values from 0.11 to 1.28 U/ml for FVIII: C, from 0.12 to 0.94 U/dL for VWF: Ag, with ratios of FVIII: C/VWF: Ag ratio above 2 after DDAVP challenge in all carriers of VWF nonsense or missense mutation. The response to DDAVP of FVIII: C and VWF parameter is normal in mild VWF deficiency related to blood group O (pseudo VWD) [1,12]. Carriers of recessive VWD type 3 heterozygous for the VWF null allele have a ratio of 2.06 for FVIII: C/VWF: Ag, and this ratio appeared to be dependent on the severity of VWD type 1 deficiency with FVIII: C/ VWF: Ag ratios of 3.2, 1.96 and 1.46 at VWF: Ag plasma levels of <30, between 30-60 and above 60 U/ml [1].
The 2008 ISTH data base reports 58 null alleles and 14 missense alleles involved in the etiology of type 3 VWD. The null alleles are located all over the VWF gene in nearly all exons 3 to 52 [27]. Missense mutations either homozygous or double heterozygous and double heterozygous for null/missense mutation as the cause severe type 3 VWD with detectable VWF after DDAVP (severe recessive VWD type 1), are mainly located in the D1-D2 domains (D47H, S85P, Y87S, D141Y, D141N, C275S, W377C, I427N), and the D4, C1 to C6, and CK domains (P2063S, C2174G, C2362F, N2546Y, C2671Y, C2754W, and C2804Y) and one in the D3 domain (C1071F). Some type 3 VWD patients who are compound heterozygous for a null allele and a missense mutation indeed detectable but very low VWF levels and measurable FVIII: C, are incorrectly diagnosed as type 3 and should to be reclassified as autosomal recessive severe type 1 VWD [14,22]. The missense mutation in carriers of 2C VWD are mainly located in the D1, D2 [1,14]. Several basic research studies have characterized congenital VWD according to the 2006 ISTH classification of VWD into recessive type 3, severe type 1, and 2N versus dominant VWD type 1 and various variant of dominant VWD IIA, IIB,IIC IIE, IID, and 2M related to domain located mutation defect on top of sensitive VWF multimeric analysis in medium to high resolution gels [28-48].
The European Canadian and UK VWD-1 studies
When correctly applying the ISTH criteria, there are several misclassifications of VWD in the European MCMDM-1VWD study (Figure 3) [53-55]. The European MCMDM-1VWD study used the VWF: RCo/Ag cut off of 0.70 for the distinction of VWD type 1 versus type 2. The European MCMDM-1VWD study did contain typical examples of recessive or heterozygous VWD type 2N (heterozygous R816W, R854W and R854W/R924Q, R854W/null). The European MCMDM-1VWD study did include typical cases of VWD 2M (D1277-E78delinsl, R1315C, R1342C, R1374C, R1374H, G1415D I1416N, Figure 3) [53-55]. There were 3 cases with typical 2M VWD with abnormal multimers and 2 mutations (R1315H/P1266L, R1315L/R934Q and R1374C/P2145S) in which the 2M mutation has a dominant negative effect on the VWD type 1 mutation (Figure 3) [53-55]. The European MCMDM-1VWD study contained mutations in exon 26, D3 domain, R1205H, R1130R/G/F, W1144G, Y1146C and C1190R (Figure 3), which usually present with a laboratory phenotype VWD 1 but do have abnormal VWF multimers with typical features of VWD 2E in the Budde VWF multimer assay [55]. Michiels classified this entity as dominant VWD type 1/2E. Such cases of VWD 1E/2E are previously classified by Eikenboom as VWD type 1, which are featured by a secretion-multimerization defect as shown in expression studies.
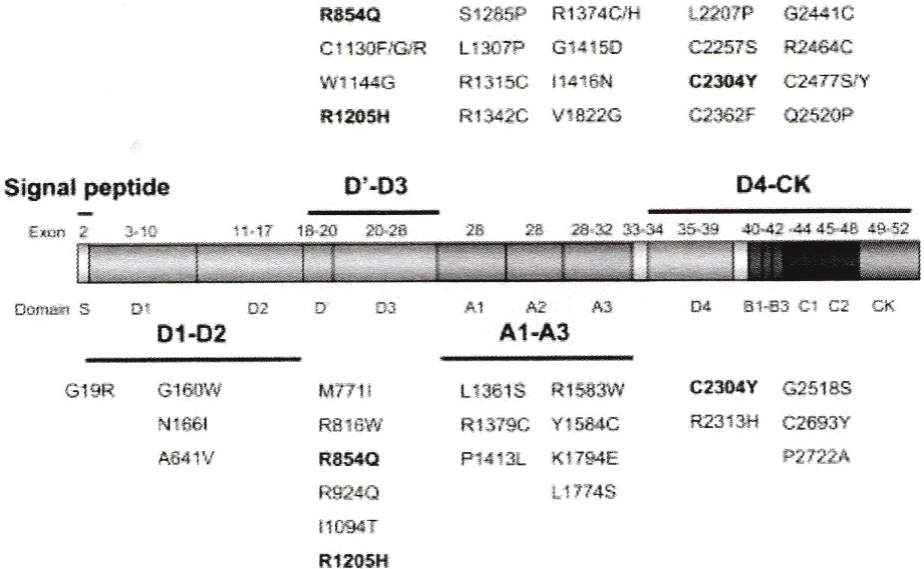
Figure 3: The spectrum von Willebrand factor gene mutations in ISTH defined type 1 Von Willebrand Disease (VWD) in the European MCDMD-1VWD study [53-
55].
The Canadian VWD type 1 study used the more stringent 0.60 cut-off for the VWF: Activity to VWF: Ag ratio as recommended by the 2006 ISTH classification to improve the distinction between type 1 and 2 VWD [13]. The Canadian study excluded the majority of VWD 2M by correct application of the SSC-ISTH criteria for 2M (Figure 4) [56]. With the exclusion of VWD type Vicenza R1205H, VWD type 1/2E, and the above mentioned cases with typical type 2N and 2M VWD, the presence of a missense mutation in patients with mild type 1 VWD and normal multimers are mainly located in the regulatory sequence region (Figure 4), the D1 D2 propeptide (pp) region (Figure 4), the D’ VWF-FVIII binding site region K762E, M771I, P812fs, Exon 21 skip, R924Q, R924W and C996E, Figure 4 [56], and only a few in the D3 (S1024fs, I1094T, Figure 4), A1 (F1280fs, R1379C, P1413L, Q1475X, Figure 6) or A2 domain (R1583W, and Y1584C, Figure 6) [53,56-59]. This large cohort of mild VWD with low to variable penetrance of bleeding, show a high (increased) prevalence of blood group O, have VWF values above 0.30 U/dL with normal ratios of VWF: RCo/Ag, VWF: CB/Ag and FVIII: C/VWF: Ag [58,59]. Within mild VWD type 1 with low penetrance of bleeding, the combination of C1584/bloodgroup O is rather frequent and typically show a good to normal response to DDAVP [58,59]. There is one case report on homozygous Y1584C mutation featured by moderate VWD type 1 with FVIII: C and VWF levels around 0.25 U/dL. The response curves of FVIII: C and VWF parameters to DDAVP in 3 studies significantly contribute to a much better characterization of patients with various variants of VWD type 1and 2 [12,13,54].
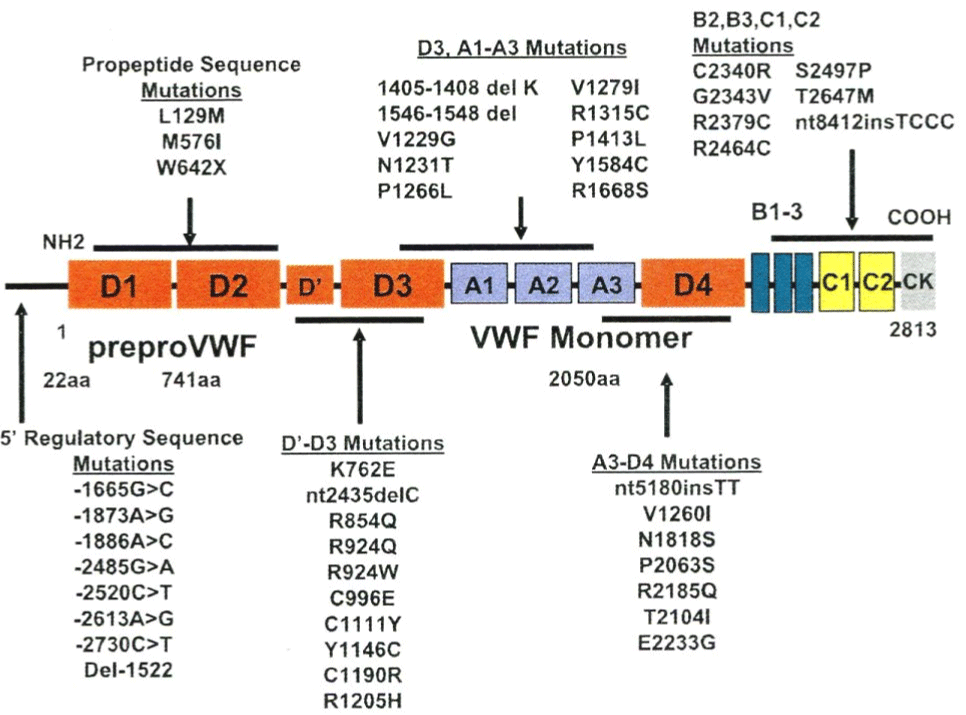
Figure 4: The spectrum von Willebrand factor gene mutations in ISTH defined type 1 von Willebrand disease (VWD) in the Canadian type 1 VWD study [56].
Figure 5 shows an update of mutations in the Canadian, European and UK VWD-1 studies [53,56-59]. Lumping the Canadian, European and UK VWD type 1 studies together the mutations 2436delC, 2685+2T>C, 3537+1G>A, 2686-1G>C 3072delC, 3108+5G>A and 3379+1G>C, Q2544X, 7437+1G>A, and 8412insTCCC are null alleles and associated with very mild VWD with low penetrance of bleeding. Carriers of a missense mutation related to severe recessive type 1 or 2C in the population are asymptomatic or manifest mild bleeding, have VWF levels around 0.50U/dL (range 0.30-0.70) 0.50 U/dl. Type 1 VWD according to the law of Mendel may become more symptomatic when associated with blood group O or another modifier of the VWF level. The Bleeding Score (BS) has also been evaluated in Obligatory Carriers (OC) of recessive type 3 VWD with a null or missense mutation [59]. From a genotypic point of view, obligatory carriers of type 3 VWD patients are similar to obligatory carriers of recessive type 1 VWD patients with a single mutated allele. Obligatory Carriers (OC) of type 3 or severe type 1 VWD with a null or missense mutation respectively may have bleeding symptoms and meet the criteria of mild type 1 VWD (Figure 6). Using the BS, Castaman et al. compared the severity of bleeding symptoms in 70 OC of recessive type 3 VWD, 42 OC of type 1 VWD and in 215 normal controls (Figure 6) [59]. OC of VWD type 3 had clearly less severe bleeding than patients diagnosed as type 1 VWD. OC of type 3 VWD with a null or missense mutation were distinct from normal controls, presenting more epistaxis, cutaneous bleeding and bleeding after surgery, further pointing to the wide heterogeneity of VWD as a heterozygous disorder (Figure 6).
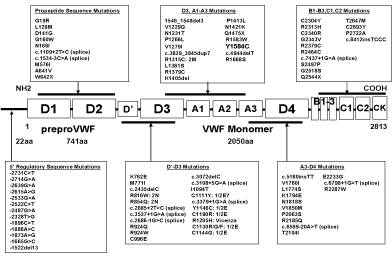
Figure 5: Von Willebrand factor gene mutation profile in the Canadian, European and United Kingdom cohorts of type 1 von Willebrand disease studies according
to Collins et al. [53,56,57-59].
The Canadian and European VWD-1 studies discovered a new category of VWD type 1 due to mutations in the D4, B1-3,C1-2 domains of the VWF gene [55,56]. Data from the European MCMDM-1VWD study show two groups of heterozygous mutations in the D4, B1-3, C1-2 domains with either Normal Multimers (NM) or Abnormal Multimers (AbM) [55]. The group with normal VWF multimers L1774S, K1794E*, C2304Y*, R2313H, G2518S*, Q2544X*, C2693Y, and P2722A have mild VWD type 1 disease, are autosomal dominant or recessive with variable penetrance of bleeding manifestations [22]. Four of these mutations in the D4, B1- 3, C1-2 domains with an increased FVIII: C/VWF: Ag ratio of 2 or more (indicated by an astrix) had complete penetrance of bleeding manifestations indicating a dominant secretion defect as the cause of VWD type 1. The group with abnormal multimers of heterozygous mutations in the D4, B1-3, C1-2 and CK domains V1822G*, L2207P*, C2257S*, C2304Y*, C2362F*, G2441C*, R2464C*, C2477Y*, C2477S*, and Q2520P* have mild to moderate VWD type 1, and usually smeary pattern of abnormal VWF multimers) [22]. Most mutations in the D4, B1-3, C1-2 and CK domain have increased FVIII: C/VWF: Ag ratios around or above 2 (indicated by an astrix). This may predict a secretion defect with restricted responses of VWF to DDAVP and some increase of clearance of VWF after DDAVP (VWD type 1 SC) [54,55]. Expression studies of mutant VWF due to mutations in the C1 to C6 domain will clarify and the mechanism abnormal banding of mutant VWF multimers as the cause of a smeary pattern in heterozygous cases due to mutations in the C1 to C6 domains [31,32]. Such smeary pattern of VWF multimers in mutant VWF are more pronounced after DDAVP [31-33].
Translation of the ISTH into the ECLM Classification of VWD 1994-2020
The splitting approach using a complete set of specific diagnostic tools and sensitive VWF assays in view of new molecular data on structure and function of VWF gene defects will be clearly superior to classify VWD compared to the lumping approach using a few insensitive tests recommended by the ISTH for the distinction of two main phenotypic variants of VWD type 1 and 2 [1-14]. Laboratory diagnosis and classification of VWD patients should be based on a complete set of laboratory measurements including bleeding time, PAF-100 closure time, FVIII: C, VWF: Ag, VWF: RCo, VWF: CB, VWFpp, RIPA, FIIII.VWF: A ratio, VWF: RCo/Ag ration, VWF: CB/ Ag ratio, VWFpp/Ag ratio and the responses of VWF and FVIII: C to DDAVP, on top of VWF multimeric pattern analysis in low, medium and high resolution gels to change or integrate the ISTH criteria into the ECLM classification of congenital VWD established ISTH criteria [1,2,10-13,28-48]. The ECLM classification is the extension of the Antwerp Classification of VWD recessive type 3, recessive type 1, recessive type 2C and 2N and sub classification of VWD type 2 into 2A, 2B, 2C, 2D, 2 1E/2E, 2 M due to mutations located in the A2, A1, D2, CK, D3 and A1 domains respectively [1,2,14]. The ECLM classification distinguished three main categories of VWD can be distinguished: first recessive type 3 and severe type 1, second dominant type 1 and 2, and third a large group of mild VWD with no or low penetrance of bleeding manifestations (Figure 7) [1,2,13,14,53-66]. In general VWD type 1 is a quantitative VWF deficiency with equally decreased values of all VWF parameters (<0.60 U/ml), a normal ratio for VWF: RCo/Ag and VWF: CB/Ag (>0.60) before and after DDAVP. VWD type 2 is a qualitative VWF deficiency with normal, near normal or decreased levels for FVIII: C and VWF: Ag and much lower values for VWF: RCo and VWF: CB with deceased ratios for VWF: RCo/Ag and VWF: CB/Ag (<0.60). VWF multimeric analysis sing low and medium resolution gels clearly distinct VWD type 2A (IIA), 2C (IIC), 1E2E (IIE) and 2D (IID) (Figures 1 and 7). The responses of FVIII and VWF parameter to intravenous DDAVP is an essential tool in the spitting approach, will clearly distinguish pseudo- VWD from true type 1 VWD, will distinguish the various variants of dominant type 1 and 2, and will elucidate differences between homozygous or compound heterozygous autosomal recessive type 3 and severe type 1 VWD [1,2]. The interpretation of VWF response curves to DDAVP has significant therapeutic implications for the different variants of recessive and dominant type 1 and type 2 VWD mutations for both clinicians and VWD patients [13,14]. Responses of FVIII: C and VWF parameters to DDAVP related to the structure and function relationship between laboratory phenotype and expression studies of VWF gene mutations significantly contribute to better understanding of the pathobiology of mutant VWF for the etiology and characterization of the various types of congenital VWD [4,9].
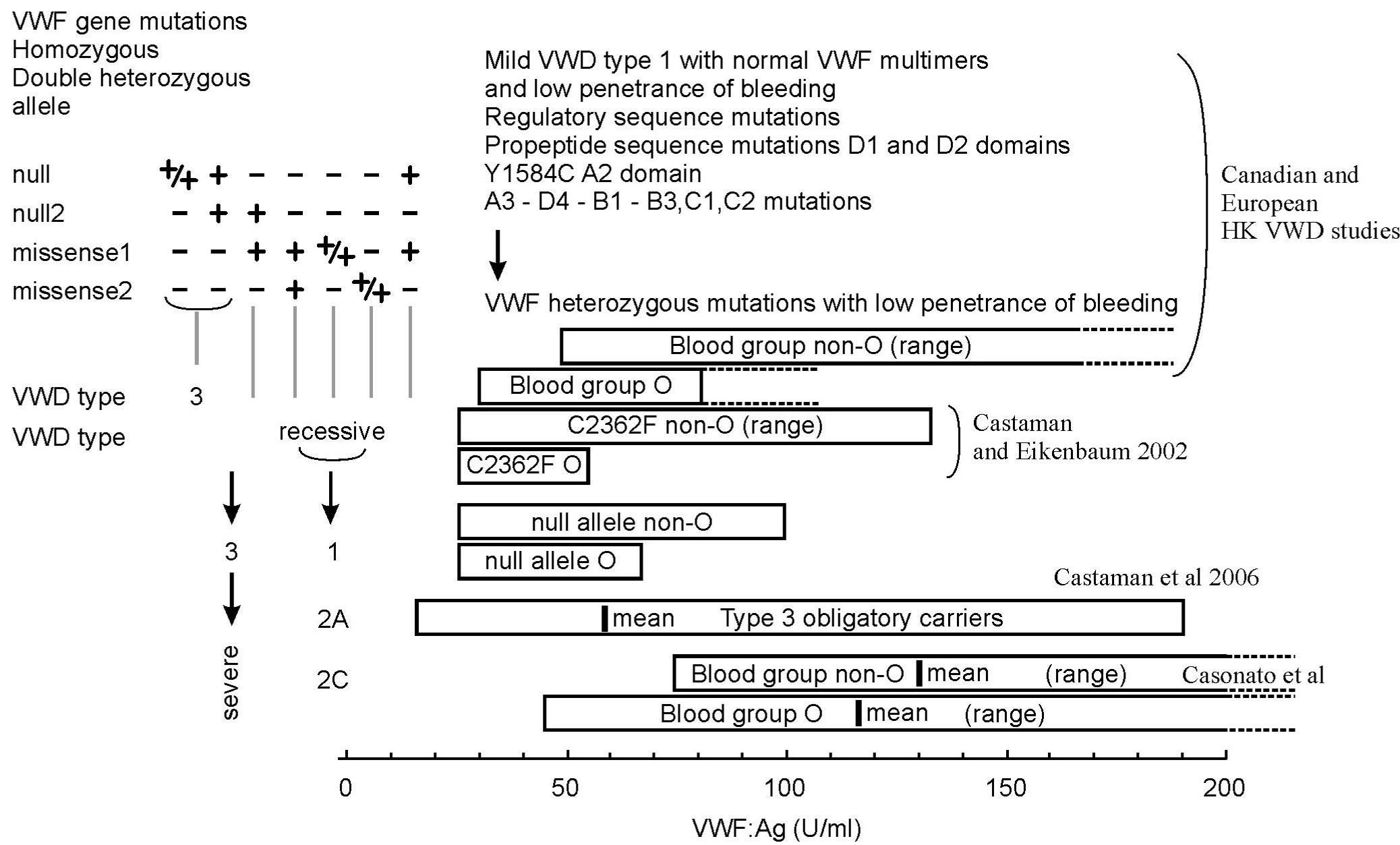
Figure 6: Relationship of LowVWF mild type 1 VWD, blood group O versus non-O, carriers of a nonsense (null) and missense allele in the VWF gene related to
recessive severe type 1 and type 3 VWD. Source Michiels et al. 2009 [59]. The 2006 VWD database of the ISTH reported 58 null alleles and 14 missense alleles
involved in the molecular etiology of recessive VWD type 3 and recessive severe type 1 VWD. Homozygous or double heterozygous null alleles as the cause
of recessive VWD type 3 are located over all domains of the VWF gene. Missense mutations either homozygous or double heterozygous or combined double
heterozygous null/missense alleles as the cause of severe recessive VWD type 1 are mainly located in the D1-D2 domains (D47H, S85P, Y87S, D141Y, D141N,
C285S, W377C and I427H) and in the D4, C1 to C6 domains (P3063S, C2174G, C2362F, N2546Y, C2671Y, C2754W and C2804Y), but not in the D3, A1 and A2
domains. VWD type compound heterozygous for null/missense alleles do have measurable FVIII and VWF after DDAVP are incorrectly diagnosed as VWD type
3 and should be reclassified as severe recessive type 1 VWD. Carriers of recessive IIC (2C) may present with LowVWF mild VWD and have mutations located in
the D1 D2 domains.
Heterozygous Obligatory Carriers (OC) of a nonsense (null) mutation related to recessive VWD type 3 and heterozygous OC of a missense mutation relate to
severe recessive type 1 VWD in the population are asymptomatic or manifest mild bleeding, have LowVWF levels around 0.50 U/dl (range 0.30-0.70 U/dL).
Following the laws of Mendel, defining recessive disease in figure 6 shows the relationship between LowVWF mild VWD type 1, blood group O versus non-O,
heterozygous Obligatory Carriers (OC) of nonsense and missense mutations in the VWF gene related to recessive sever type 3 and type 1 VWD. OC of null allele
or a missense allele do have lower VWF values than non-carriers, OC of a null or missense allele may become symptomatic when associated with blood group
O. (Castaman et al. reviewed by Michiels [59]). From a genomic point of view OC of VWD type 3 and severe recessive type 1 are very similar and both do have
restricted responses of VWF as compared to FVIII: C after DDAVP with FVIII: C/VWF: Ag ratios around 2 after DDVP. Using Bleeding Score (BS) assessment,
The data of Castaman in figure 6 compared the bleeding severity in 70 OC of recessive VWD type 3, in 42 OC of recessive severe type 1 VWD. OC carrying a nul
allele of VWD type 3 had less severe bleeding than OC carrying a missense mutation of recessive type 1 VWD. OC carrying a missense mutation were distinct from
controls and presented more epistaxis, cutaneous bleeding and usually did not significantly bleed after surgery [59].
The concept in figure 6 point to the wide heterogeneity of LowVWF mild type 1 VWD patients. LowVWF mild VWD indeed is a heterogenous disorder with variable
penetrance of mild mucocutaneous bleeding manifestations and slight prolongation of PFA-CT [59]. LowVWF mild type 1 VWD patients are clearly distinct from
dominant variant of VWD type 1 SD, 1C Vincenza, 1E/2E (IIE) and 2M featured by bleeding since early childhood, high Bleedings Scores (BS) and strongly
prolonged PFA-CT during life-long follow-up.
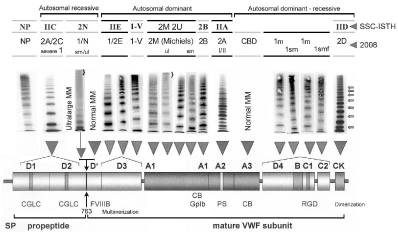
Figure 7: Clustered distribution of von Willebrand factor (VWF) gene mutations, diagnosis and classification of Von Willebrand Disease (VWD) type 1 and VWD
type 2 with abnormal multimers: comparing the SSC-ISTH and the proposed ECLM classification in 2008 by Gadisseur & Michiels [65].
The role of FVIII: C/VWF: Ag ratio in the ECLM classification of VWD
By definition, the concentration in plasma of FVIII: C and VWF: Ag is 1.0 U/dL [13]. Consequently, the ratio FVIII: C/VWF: Ag is around 1 in normal individuals with blood group O and non-O [14,59]. The ratio of FVIII: C binding sites on VWF: Ag on a molecular basis is 1: 50 and independent of the size of VWF multimers indicating that many potential FVIII: C binding sites on VWF: AG are free. As the VWF: Ag is 50% of normal due to decreased biosynthesis in quantitative VWD type 1 due to a secretion or synthesis defect, the ratio of FVIII: C/VWF: Ag will increase to about 2 [14,61]. Carriers of recessive VWD type 3 heterozygous for the VWF null allele (decreased synthesis) have a ratio of 2.06 for FVIII: C/VWF: Ag, and this ratio appeared to be dependent on the severity of the VWF: Ag deficiency with ratios of 3.2, 1.96 and 1.46 at VWF: Ag plasma levels of <30, between 30-60 and above 60 U/ml [14,61]. An increased FVIII: C/ VWF: Ag ratio in VWD type 1 and 2 refers to a VWF secretion defect of mutated VWF as can be documented by a restricted response of VWF to DDAVP and a normal response of FVIII: C to DDAVP [12]. An increased FVIIIC/VWF: Ag ratio, a poor response of VWF: Ag as compared to a restricted response of FVIII: C and asymptomatic parents is typically seen in severe recessive type 1 or 2 VWD. A normal FVIII: C/VWF: Ag ratio is consistent with normal secretion of mutant VWF protein in type 1 (including Vicenza) and in type 2 (2A group II and 2B) VWD patients [2,12,61]. Short half life time of VWF parameters after DDAVP in type 1, Vicenza, 1E2E (IIE), 2M and 2U indicates rapid clearance not due to proteolysis [13,14]. Short half life times of functional VWF parameters in type 2A (IIA) and 2B (IIB) indicates increased proteolysis [2,12,14,61,64]. A decreased FVIII: C/ VWF: Ag ratio (less than 0,50) due to a FVIII: C binding defect of the VWF protein plus recessive inheritance refers homozygous or double heterozygous type 2N VWD [17-20]. A decreased FVIII: C/VWF: Ag ratio is also seen in heterozygous mild VWD Normandy and in mild hemophilia A due to a VWF binding defect in the FVIII gene [16-20]. A normal response of VWF parameters and restricted response of FVIII: C to DDAVP followed by shortened half life times of FVIII: C refers to VWD 2N due to a FVIII binding defect in the VWF or mild hemophilia due to a VWF binding defect in the FVIII protein [17,19,20].
Autosomal recessive VWD
The inheritance of VWD type 3 is autosomal recessive [21-26]. Type 3 VWD with virtual complete VWF deficiency are homozygous or compound heterozygous for two null alleles (gene deletions, stop codons, frame shift mutations, splice site mutations, and absence of MRNA) in the majority and rarely compound heterozygous for a null allele and a missense mutation or homozygous for a missense mutation [27]. Compound heterozygosity for a null allele and a missense mutation or homozygosity or double heterozygosity for missense mutations is not consistent with type 3 VWD, but common in patients with severe autosomal recessive type 1 VWD [28-34]. Autosomal recessive severe type 1 VWD patients have low VWF and FVIII: C levels may range from 0.01 U/dL up to around 0.30 U/dL. A considerable number of missense mutations related to autosomal recessive severe type 1 VWD have been identified in the VWF prosequence (D1 and D2 domains), in the C1 to C6 domain and the dimerization site (CK domain) [28-34]. Autosomal recessive VWD type 2C (IIC) is rare and caused by homozygosity for a missense mutation or double heterozygosity of a null allele and missense mutation in the D1 and D2 domains (exon 11 to 16) of the VWF propeptide (Figure 4) [35-41]. Autosomal dominant or recessive VWD 2D is caused by missense mutations in the CK domain (Figure 4) [34].
Von Willebrand Disease Normandy (VWD 2N) due gene mutations in the factor VIII binding domain (D’-D3, Figure 4) typically featured by reduced FVIII: C levels despite normal or near normal VWF: Ag, VWF: RCo and VWF: CB levels, normal VWF: RCo/Ag ratio, and a normal VWF multimeric pattern as well as normal VWF dependent platelet functions including RIPA and bleeding time (VWD type 1/N [16-20]. Recessive VWD type Normandy is frequently misclassified as mild hemophilia. Intravenous DDAVP in VWD type Normandy and in mild hemophilia A due to a VWF binding defect of FVIII show completely normal responses for VWF parameters consistent with type 1 VWD, but restricted responses of FVIII: C followed by shortened half life times [17,19]. The degree of restricted response of FVIII: C to DDAVP depends on the severity the FVIII binding to VWF [17]. In a large group of 14 unrelated patients with mild FVIII: C deficiency and normal values for VWF 15 were diagnosed as homozygous VWD type 2N (FVIII: C 0.21±14 U/ dl, VWF: Ag 0.79±0.43 U/dl) and 5 were diagnosed as heterozygous VWD 2N for (FVIII: C 0.32±06 U/dL, VWF: Ag 0.91±0.18 U/dl, and VWD type 2N could be excluded in 124 (FVIII: C 0.29±0.12 U/dl, VWF: Ag 1.03.3±0.45 U/dL) [18]. The 15 type 2 Normandy patients were either homozygous or compound heterozygous for known mutations whereas the 5 individuals with intermediate VWF: VIIIB values were heterozygous for the R854Q type Normandy mutation [18].
Autosomal dominant VWD
Lumping of dominant IIA (2A), recessive IIC (2C), dominant IIE (1E/2E), and dominant or recessive IID (2C) as type 2A by the SSC-ISTH (Figure 4) is illogical and confounding. The type 2 VWD phenotypes IIA (2A) and IIB (2B) are clearly different from VWD types IIC (2C), IID (2D) and IIE (2E) (Figures 1 and 7) [14]. Careful analysis of reported cases with dominant VWD type 1 due to mutations in the D3 domain (multimerization-secretion-clearance defect) are featured by a type 2E multimeric pattern in medium resolution gels (Figures 1,7 and 8) [42-48]. Budde & Schneppenheim proposed in 2001 and 2005 a novel molecular classification of type 1 and 2A VWD with abnormal multimers (2A, 2B, 2C, 2D, 2E, 2-Normandy in Figure 4 versus VWD type 1/2E, 2M or 2U, and type 1sm/2M in Figures 7 and 8) mainly based on current knowledge regarding the structure and function relationship of the normal and mutated VWF gene and proteins [4,10,49-52]. By applying a complete set of laboratory tools including a sensitive method for the analysis of the VWF multimeric pattern within the setting of the European study on Molecular and Clinical Markers for the Diagnosis and Management of Type 1 VWD (MCMDM-1VWD), Budde & Schneppenheim produced consistent good data on the relation between VWF gene mutations and the proposed classification of dominant VWD type 1 and 2 in Figures 1,7 and 8 [4,10,49-52].
In his review on VWD 2N Michiels draw attention to the fact that carriers of missense mutations R763Q, R760C, Y795C located in or around the VWF propeptide (pp) cleavage site 763 have ultralarge VWF multimers with a smeary (sm) pattern also on top of VWD type Normandy when associated with R854Q or null allele. Ultralarge VWF multimers are also seen in VWD Vicenza R1205H/M740I in after DDAVP or exercise, but such ultralarge VWF multimers are never seen in VWD type 1E/2E (Figure 8) [53-55]. LowVWF mild VWD patients are characterized by low to variable penetrance of bleeding when associated with blood group O (Figure 6) [56]. The lowest values of VWF in LowVWF mild VWD type 1 are around 0.30 u/dL with normal ratios for vWF: RCo/Ag and VWF;CB/Ag. (Michiels et al 2009, Figure 6) [56]. About half of patients diagnosed as type 1 in the European MCMDM-1 study had abnormal VWF multimers, which were diagnosed as VWD type 2E in the D3 domain, 2M, 2A-like (2U) in the A1 domain, and type 1 with a smeary pattern of VWF multimers in the D4, B1-3, C1-2 domain (Figure 8) [53-55]. Mild VWD type 1 due heterozygous mutations of the D1 and D2 usually decreased values for VWFpp and normal VWF multimeric pattern in a low resolution gel [54]. All mutations in the FVIII binding domain show a multimers normal VWF multimeric pattern except due to noncysteine 2N mutations in the D3 domain. Interestingly, homozygous cysteine mutations C788R, and C1225G and double heterozygous C788T/null show a hybrid VWD phenotype of pronounced severe VWD 2E with factor VIII binding defect of about 50% (VWD 2E/N). VWD patients with mutations in the D3 (multimerization) domain (C1130R, C1130G, W1144G, Y1146C, C1190F) frequently show a type 1 VWD laboratory phenotype but type 2E pattern with relative loss of large VWF multimers and reduced triplet structure and proteolysis (Figure 8). Loss of function mutations in the A1 domain (L1307P, R1315C, R1315L, R1374C, R1374H, G1415D) show the presence but relative loss of large VWF multimers in medium resolution gels, no increase of triplet structure, and a defect of the ristocetine cofactor activity and RIPA consistent with VWD type 2M or 2U (Figure 8) [4,10,12,14,65]. Several mutations (L2207P, C2257S, C2304Y, R2379, G2441C, R2464C, R2464C, C2469P, C2671Y) in the D4, B1-3, C1-2 domain show a dominant type 1 VWD phenotype show an atypical smeary pattern of large multimers and no triplet structure of small multimers (Figure 8) [55].

Figure 8: In the European MCMDM-1VWD study VWF multimeric pattern in VWD 1E/2E(IIE) is featured by the loss of large multimers and each of the VWF bands
lack the triplet structure seen in VWD IIA and IIB (figure 1) [55]. VWD 2M is featured by the a smeary pattern of large and intermediate VWF multimers, VWD 2U
or 2A-variant reveals some loss of large VWF multimers and the triplet structure of each VWF band is less pronounced as compared to VWF from VWD IIA (2A)
and IIB (2B) patients (figure 7) [55]. In the European MCMDM-1VWD study, Dr Budde observed that in 6 of eight mutations L2207P, C2257S, C2304Y, C2362F,
C2441Y, C2441 C, R2464C, C2477Y, C2477S in exon 38, 40, 42, and 43 (D4, B1-B3 C1 domain) are clearly VWD type 1 showing a smeary VWF multimeic pattern
indicated as sm or smf with the presence of large VWF multimers and a laboratory phenotype of mild to moderate VWD type 1 [55]. The responsible mutations with
a smeary VWF pattern in the D4, C1 to C6 domain were cysteine (C) mutations in 6 of these 8 mutations [55].
Data from the European MCMDM-1VWD study show that the heterozygous mutations in the D4, B1-3, and C1-2 domains (L1774S*, K1794E*, C2304Y*, R2313H, G2518S*, Q2544X*, C2693Y*, and P2722A) have normal multimers and have mild VWD type 1 disease with variable penetrance of bleeding manifestations. Heterozygous mutations in the D4, B1-3, C1-2 and CK domains V1822G*, L2207P*, C2257S*, C2304Y*, C2362F*, G2441C*, R2464C*, C2477Y*, C2477S*, and Q2520P* have mild to moderate VWD type 1, abnormal VWF multimers (usually smeary pattern) [53,55]. Nearly all mutations in the D4, B1-3, C1-2 and CK domain have increased FVIII: C/VWF: Ag ratio around or above 2 (indicated by an astrix*) indicating a Secretion (S) defect [61], which predict restricted response of VWF to DDAVP and more or less rapid Clearance (C) after DDAVP (VWD type 1 SC). Expression studies of mutant VWF are predicted to show abnormal banding of VWF multimers as the cause of a smeary pattern, which is more pronounced after DDAVP [54,55].
The role of VWF multimeric pattern and VWF-propeptide in the characterization of VWD type 1 and type 2
Haberichter et al in the USA [47,63], Casonato in Italy66 and the European MCDMDM-1VWD study [53-55] introduced the role of VWF-propeptide in the characterization of VWD (Table 4, Figure 9 and 10). The VWF: pp and VWF: Ag are non-covalently associated and stored in Weibel-Palade bodies in endothelial cells for regulated release [60-62]. After release in plasma, VWFpp and mature VWF multimers dissociate and circulate independently with a half- life of 2 to 3 hours for VWF: pp and a half-life time of 8-12 hours for VWF: Ag. Concentrations of VWF: pp and VWF: Ag in plasma are expressed in units per ml of normal plasma, consequently, the ratio of VWF: pp to VWF: Ag in plasma is defined to equal 1.0. The rationale behind the use of the VWF: pp/VWF: Ag ratio as an indicator for increased clearance of VWF is that in all variants of VWD type 1 and 2 the half life time of VWF: pp is normal, whereas the clearance of VWF: Ag may be very short, shortened or normal. In patients with VWD type 1 and type2 VWD patients with a secretion defect but normal clearance do show a restricted response of VWF to DDAVP followed by normal half lifetimes of VWF: Ag are expected to have decreased values for VWF: pp and VWF: Ag with normal ratio for VWF: pp/VWF: Ag. VWD patients with a normal secretion but increased clearance of VWF: Ag plus VWF: RCo and FVIII: C do show good responses to DDAVP followed by short half-life times of VWF: Ag which result in increased VWF: pp/VWF: Ag ratio. Four original studies of Haberichter et al. [47,63], MCMDM-1VWD study [54] and Davies et al. [64] analysed the results on VWF: pp and VWF: Ag related to clearance of VWF: Ag after DDAVP as a component in the etiology of type 1 and type 2 VWD (Table 5, Figures 9 and 10). Davies et al analysed the findings in mild VWD related to the C1584 mutation in comparison with controls blood group O vs Non-O [64]. Table 5 summarizes the results of VWF: pp/Ag ratios related to the level of VWF: Ag and VWF: Ag survival times after DDAVP in VWD type 1/2E, and type 1 Vicenza due to mutations in the D3 domain, VWD type 1SC due to a mutation in the D4 domain and mild VWD type 1 due to mutations in the D1-D2-D’ domains, the D4-B1-B3- C1-C2 domains and the C1584 mutation with blood group O.
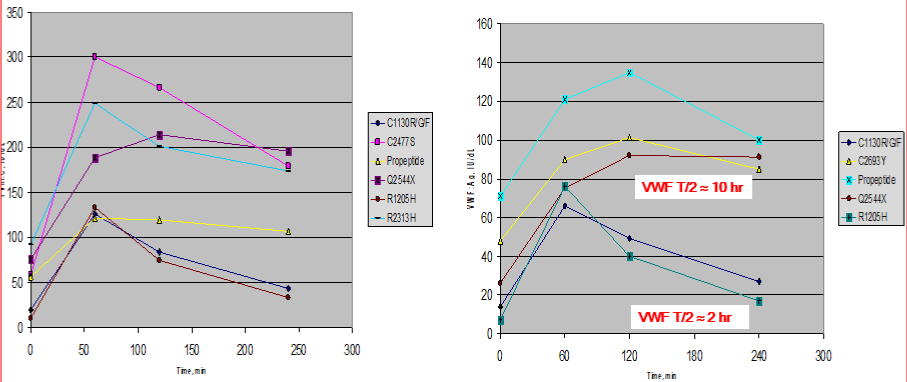
Figure 9: Responses of FVIII;C (left) and VWF: Ag (right) after intravenous DDAVP in VWD patients with the mutation R1205H and C1330F/G/F in the D3 domain,
and the mutations C2477S, Q2544X, R2313H, C2693Y, and Q2544 in the C1 to C6 domains or a mutation in the propeptide (pp) domain D1 D2 domain. Please
note that the response of FVIII;C is 2x higher than of VWF: Ag. The clearance of FVIII/VWF: Ag is increased in the D3 mutations R1205H and C1130F/G/F (half
life time of FVIII;C/VWF: Ag about 2 hours), whereas the FVIII/VWF: Ag half-life times are nirmal around 10 hours in VWF mutations in the D1, D2, C1 to C1 to C6
domains of the VWF gene. These findings are consistent with the results shown in table 4. Source Castaman et al. [54].
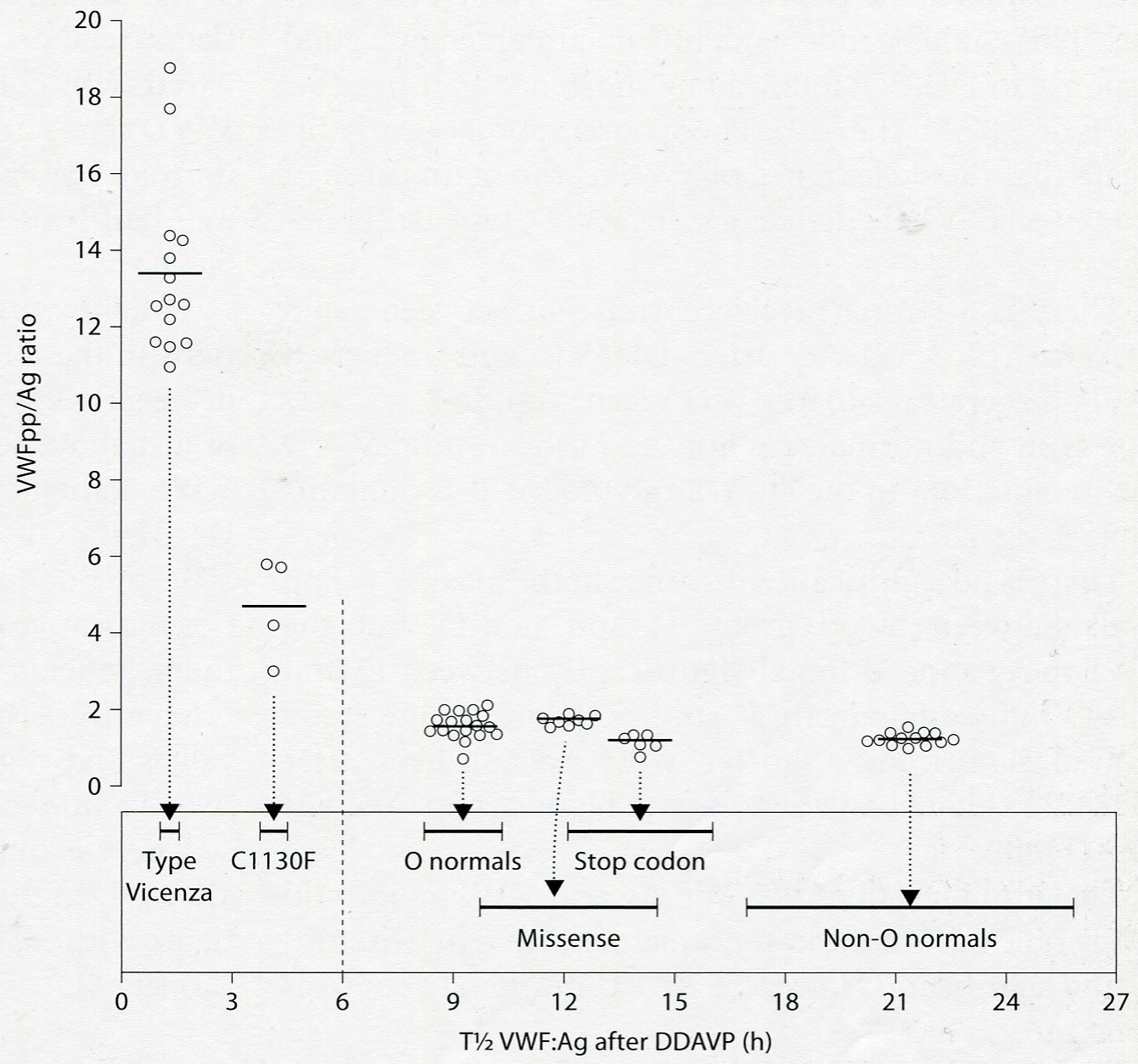
Figure 10: Result of VWFpp/Ag ratio versus half life time of VWF: Ag after DDAVP in the Italian study in patients with VWD type Vicenza (R1205H), VWD type
1E/2E (C1130F) and in LowVWF type 1 mild VWD due to stop codon, nonsense mutation, and in controls blood group O vs non-O [66].
![]()
VWD type
Number
VWF:Ag
VWF:pp
VWF:
T1/2 VWF:Ag
Mutations
patients
IU/dl
IU/dl
pp/Ag
after DDAVP
Mild type 1
D1, D2, D’ domain
G160W, N116I, M771I
6
46.1
64
1.13
near normal
R854Q/R854Q
37-79
23-96
0.6-1.6
to normal
Type 1/2E (IIE)
W1144G
6
16
61
4.1
2 to 4 hours
Blood group O
13-22
55-69
3.1-5.1
Type 1/2E (IIE)
W1144G
6
22
72
3.2
2 to 4 hours
Blood group non-O
14-32
68-80
2.1-5.0
Type 1 SC
S2179F
5
15
65
4.8
2 to 4 hours
Blood group O
01-Sep
56-77
3.2-6.4
Type 1 Vicenza
R1205H/M740I
3
6.8
72.8
10.9
less than 2hrs
5.7
63-85
10.1-11.5
Mild type 1
A3, D4, B-C domains
6
58.4
77.6
1.85
near normal
28-65
66-109
1.0-2.8
to normal
Davies et al [62]
C1584 all
50
82 ± 35
94 ± 20
1.36 ± 0.40
-
C1584 group O
23
58 ± 14
94 ± 20
1.66 ± 0.33
-
C1584 group non-O
17
98 ± 34
109 ± 30
1.17 ± 0.25
-
Blood group O
50
97 ± 24
110 ± 31
1.16 ± 0.24
-
Blood group non-O
50
126 ± 30
110 ± 28
0.90 ± 0.21
-
Table 4: Result of VWF: Ag and VWF: pp levels in VWD type 1/2E or 1 SC with the mutations W1144G (D3 domain) and S2179F (D4 Domain) and blood group O and in mild VWD type 1 due to mutations in the D1-D2-D’ domain and the D4-C1-C3-C1-C2 domain in the studies of Haberichter et al. [47,63] as compared to C1584 [64] and controls related to ABO blood group.
![]()
Mutation/
FVIII:C
VWF:Ag
VWF:RCo
VWF:CB
RCo/Ag
CB/Ag
VIII:C/Ag
VWD type
Domain
U/dL
U/dL
U/dL
U/dL
ratio
ratio
ratio
L2270P/C1
0.71
0.26
0.19
0.35
0.73
1.35
2.7
1 SD sm
C2257S/C1
0.70
0.29
0.42
0.30
1.45
1.03
2.4
1 SD sm
C2304Y/C1
0.42
0.20
0.19
0.37
0.95
1.85
2.1
1 SD sm
C2362F/C2
0.88
0.35
0.34
0.39
0.97
1.11
2.5
Mild 1 SD sm
G2441C/C3
0.61
0.22
0.20
0.26
0.91
1.18
2.8
1 SD sm
C2464C/C3
0.32
0.25
0.25
0.37
1.00
1.48
1.7
1 SD sm
C2464C/C3
0.52
0.33
0.30
0.50
0.91
1.52
1.7
Mild 1 SD sm
C2477Y/C4
0.68
0.38
0.33
0.43
0.87
1.13
1.8
Mild 1 SD sm
C2477S/C4
0.58
0.28
0.40
0.31
1.43
1.11
2.1
1 SD sm
Q2520P/C4
0.48
0.19
0.27
0.19
1.42
1.0o
2.5
1 SD sm
VWD type 1 with normal VWF multimers due to mutations in the D4-C1-6 domain in the European MCMDM-1VWD study include L1774S, K1794E*, C2304Y*, R2313H, G2518S*, Q2544X*, C2693Y, and P2722A, have mild VWD type 1 disease, are autosomal dominant or mild with variable penetrance of bleeding manifestations. The laboratory features of the VWD type 1 VWD patients with abnormal multimers of heterozygous mutations in the D4 and C1-6 CK domains shown in table V1822G*, L2207P*, C2257S*, C2304Y*, C2362F*, G2441C*, R2464C*, C2477Y*, C2477S*, and Q2520P* have mild to moderate VWD type 1, and a smeary pattern of abnormal VWF multimers (sm). The majority of the mutations in the D4 and C1-6 have increased FVIII:C/VWF:Ag ratios around or above 2 (indicated by an astrix) indicating a secretion defect and a poor or restricted response of VWF parameters to DDAVP.
Table 5: VWD type 1 SD and smeary WF multimers due to heterozygous mutations in the C1 to C6 domains in the European MCMDM-1VWD study [55].
The VWF: pp/Ag ratios in two studies of Haberichter et al are increased cases of VWD type 1/2E with the mutation C1130, W1144G in the D3 domain and in case S2179F type 1 mutation in the D4 domain indicating that a pronounced increased clearance (C) of the VWF: Ag [47,63]. This could be documented by a good response to DDAVP of VWF: Ag followed by shortened half-life times for VWF: Ag of about 2 to 4 hours as demonstrated by Casonato in the Italian study (Figure 9) [65,66]. These observations clearly indicate that rapid clearance of VWF: Ag also contributes pronounced VWF deficiency in VWD type 1/2E (secretion clearance defect, Figure 9).There is a strong inverse correlation between rapid clearance of VWF: Ag and increased VWF: pp/Ag ratios above 2 seen in VWD type 1/2E. Increased clearance of VWF is not in mild VWD due to mutations in the D1 D2 and the D4-B-C domains (Table 4) [63-66]. The response to DDAVP in VWD Vicenza (1C) is very good for FVIII: C, VWF: Ag and VWF: RCo, which is followed by very short half-life times of less than a few hours for FVIII: C and all VWF parameters indicating a rapid clearance defect of VWF: Ag as the explanation of very high VWF: pp/VWF: Ag ratios above 10 (Table 4, Figure 4&10) [63-66]. The quantitative response of FVIII and VWF parameters to DDAVP in VWD Vicenza is normal indicating normal secretion, which is followed by rapid clearance of VWF: Ag resulting in very high VWF: pp.Ag ratios. Patients with VWD type 1/2E (IIE) due to C1130G/For R and W1144G mutations in the D3 multimerization domain and the S2179F mutation in the D4 domain have increased VWFpp/Ag ratios despite decreased or low absolute values for VWF: pp (Table 4 Figures 9 and 10) and shortened VWF: Ag half-life times indicating rapid clearance as a main mechanism for a laboratory phenotype 1. Patients with mild VWD type 1 due to mutations in the D1, D2 and D’ domains have decreased VWF: pp levels and normal VWF: pp/Ag ratios of just above 1 indicating for a mild secretion defect of VWF [63-66]. Patients with mild VWD due to mutations in the D1 D2 domains and in the D4-B1-B3-C1-C2 domains have decreased values for VWF: pp but slight increased VWF: pp/Ag ratios indicative for a secretion defect (high FVIII: C/VWF: Ag ration) and no clearance defect (Table 4) [63]. Patients with the C1584 mutation and blood group O have normal values for VWF: pp, near normal VWF;: pp/ Ag ratios indicative for a mild clearance defect of VWF. Patients with recessive severe type 1 VWD are predicted to have decreased values for both VWF: Ag and VWF: pp and normal VWF: pp/Ag ratios reflecting a secretion defect (VWD type1SD) [54,66].
Molecular etiology and pathology of VWF behind the ISTH and ECLM classifications
With the advent of a complete of new rapid and specific VWF assays and VWF multimer analysis Katarzina, Smejkal & Vangenechten induced an earth breaking undermining of the ISTH criteria as an artificial authorative classification by dictation based on a few insensitive test not based on basic research and without scientific evidence [67-70]. Katarzina, Smejkal & Vangenechten in fact presented a novel focus on the molecular basis of VWD genotype-phenotype classification behind the ISTH and the ECLM classifications [67-70]. Basic insights into the molecular etiology and pathology of VWF and related clinical phenotypes start with the detection of domain located mutation in the D1, D2, D’, D3, A1, A2, A3, D4, C1 to C6 and CK domain of the VWF gene followed by phenotyping of each individual VWD patient with a complete new set of PFA-CT, FVIII: C, VWF: Ag, VWF: RCo (HemosIL), VWF;GPIbM, VWFpp, assays RIPA, VWF: RCo/Ag,VWF;CB/Ag, VWFpp/Ag VWF multimers in a low, medium or high resolution gel on top of a DDAVP challenge test [70]. Katarzina, Smejkal & Vangenechten directly compared the performances of the rapid VWF assays VWF.GPIbR and VWF;GPIbM and VWFpp against a complete set of classical VWF assays VWF;Ag, VWF: CB, RIPA on top of VWF multimeric analysis in a low, medium and high resolution gel in the large Brno cohort of VWD patients VWD type 1, 2N,andtype 2 due to mutations in the D1, D2, D’, and D3 domains of the VWF genes [67,68]. The ECLM phenotype of VWD type 1 with heterozygous mutations in the D1 domain (Brno cohort, Smejkal Table 5) [68] is clearly distinct from VWD type 1M and type 1Msm caused by heterozygous mutations in the D4 and C1 to C6 domain as described by Budde in the European MCMDM-1VWD study [55]. VWD patients due to mutations in the D3 domain are of type 1E or 2E multimerization defect associated with an additional secretion defect (SD, increased FVII: C/VWF: Ag ratio) and or an additional clearance defect (1C Vincena, or W1144G VWD 1E C SD) defect (Table 6). The combined use of VWF;Ag, VWF: RCo (HemosIL), VWF: CB and VWF GPIbM and RIPA on top of low medium and high resolution VWF multimer analysis in need to make a correct diagnosis of VWD 2BC, VWD 2B, VWD 2M VWD 1M [69]. The P1266L mutation refers to VWF 1M VWD Malmo or New York. Heterozygous mutations in the A3 domain has been described as VWD 1m with selective VWF Collagen Binding Defect (VWFCBD). The spectrum of VWD type 1m or type 1msm due to mutations in the D4 and C1 to C6 domains are not yet clearly defined. A detailed diagnostic basic research approach in defining each individual VWD patient using a complete nover set of sensitive VWF assays is mandatory to realise the scientific goal of precision and personalized medicine management options in all variants of recessive and dominant type 1, type 2 and type 3 VWD patients [67-70].
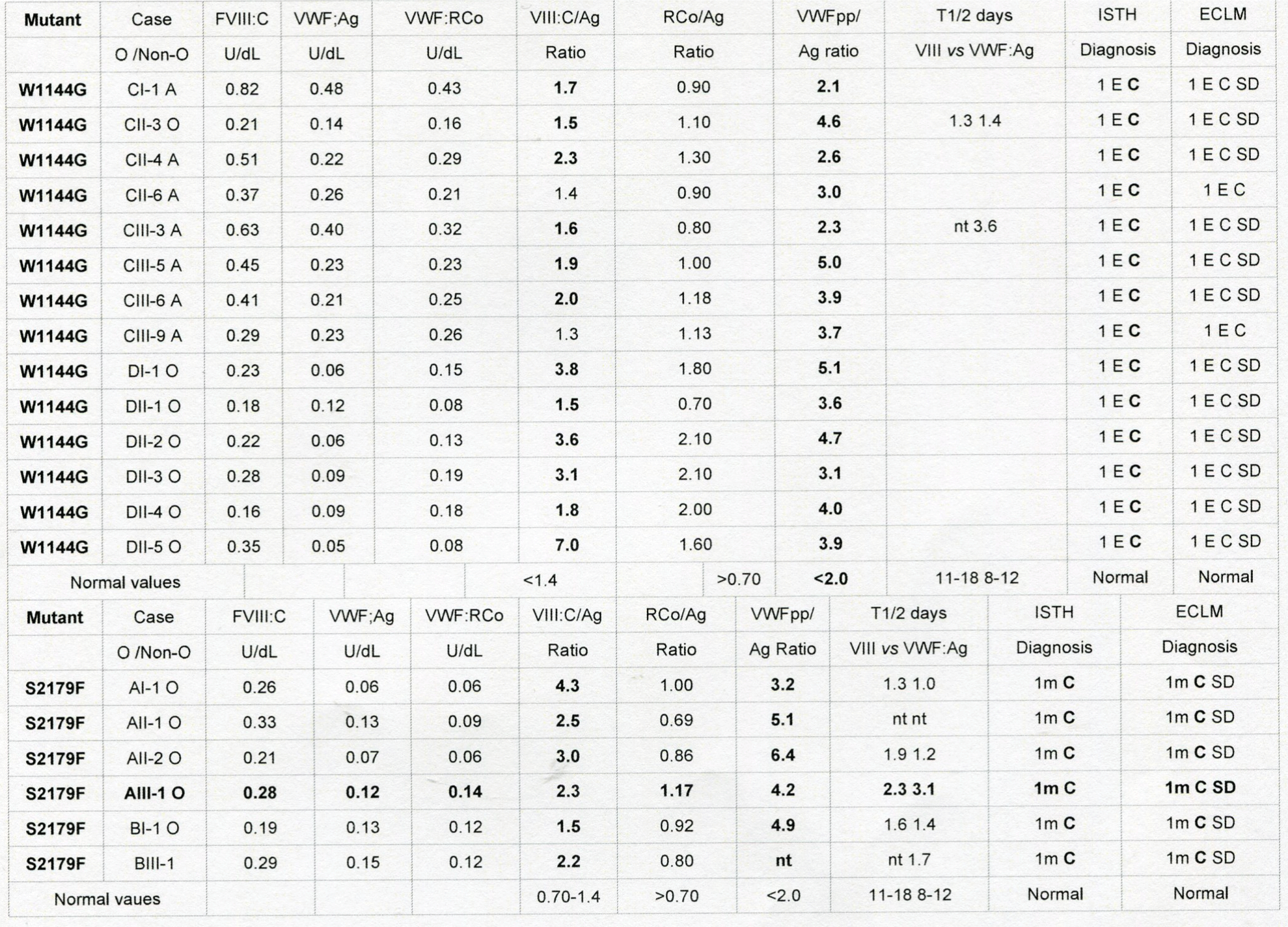
table 6: Critical analysis according to the ISTH compared to the ECLM classification of VWD by the combined use of a complete set of VWF assays FVIII: C, VWF: Ag, VWF: RCo and VWFpp of dominant VWD type 1 mutation W1144G (ISTH) (Haberichter et al.) [47,63] in the D3 domain and dominant VWD type 1 mutation S2179F (ISTH) (Haberichter et al)47,63 in the D4 domain. Subsequent ECLM reclassification of the W1144G mutation changed the diagnosis into VWD 1E C SD clearance and secretion defect by application of VWF: pp/Ag ratio and FVIII: C/VWF: Ag ratio and changed the S2179F mutation into VWD 1m C SD clearance and secretion defect by application of VWF: pp/Ag ratio and FVIII: C/VWF: Ag ratio.
The role of PFA-CT in the diagnostic work-up of patients with von Willebrand disease and controls has recently been evaluated by Nummi et al by measuring PFA-CT using Epinephrin (EPI) and Adenosinediphosphate (ADP) cartridges in a one center cohort of 54 ISTH defined VWD patients: LowVWF type 1 VWD in 10, classical type 1 in 7, type 2A in 14, type 2B in 9, type 2N in 1, type 3 in 13 and normal VWF in no VWD 19 (Figure 11) [71]. The VWF: RCo and VWF: Ag levels were between 0.14-0.33 U/dL and 0.08-0.25 iU/dL in classical dominant VWD type 1 and between 0.43-0.64 U/dL and 0.36-0.64 in LowVWF mild type 1 VWD respectively. PFA-CT EPI and ADP were normal (less than 150 seconds in healthy controls and in the group of no VWD with normal VWF levels. The PFA CT EPI and ADP were prolonged with values between 150-250 seconds in 6 of 10 and in 2 of 10 LowVWF mild 1 VWD patients. The PFA CT EPI and ADP were strongly prolonged above 250 seconds in VWD 2A, 2B, 2M and type 3. The PFA CT EPI and ADP prologations in mild VWD type 1 are in between the values seen in LowVWF versus VWD type 2A, 2B and 2M (Figure 11) a normal PFA-CT in LowVWF mild VWD type 1 excludes a clinical relevant bleeding tendency [71].
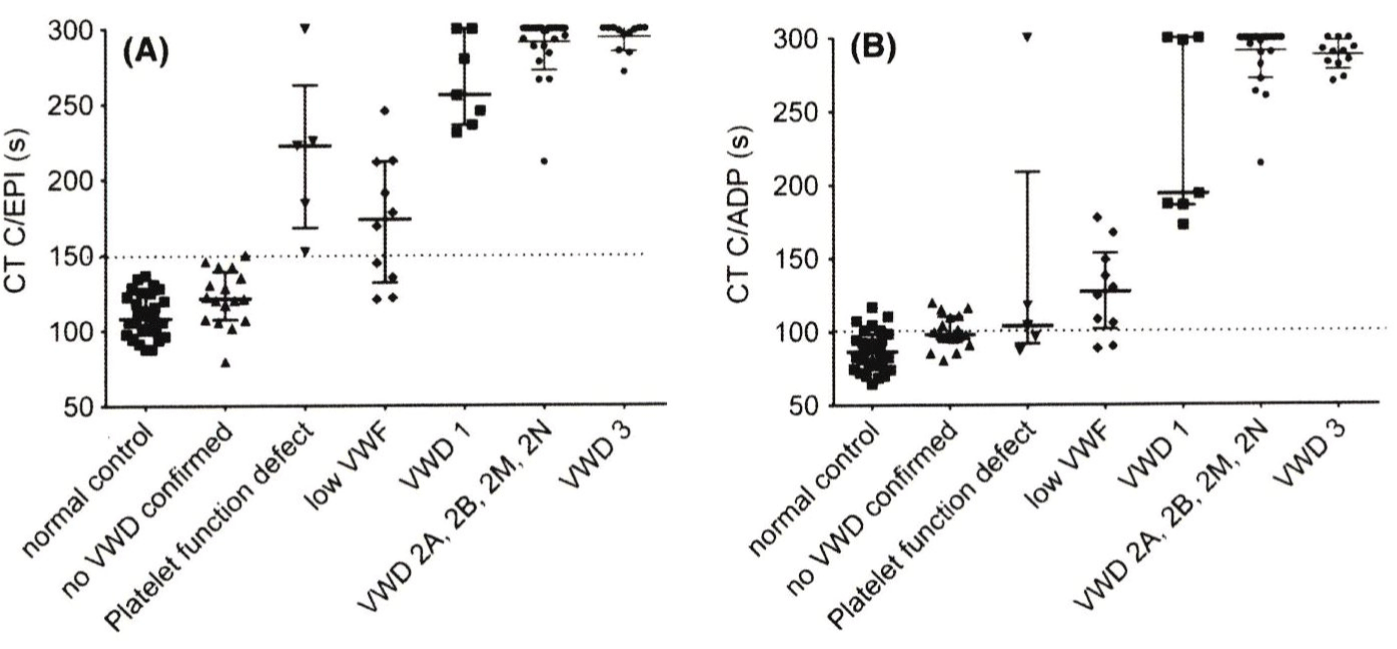
Figure 11: Results of Platelet Factor Analyzer Closure Times (PFA-CT) with Epinephrin (EPI) and Adenosine Diphosphate (ADP) cartridges in a one center study
of Nummi et al in normal controls, individuals with no VWD and in 54 ISTH defined VWD patients: LowVWF in 10, type 1 in 7, type 2A in 14, type 2B in 9, type 2N
in 1, type 3 in 13 and normal VWF in no VWD 19 [33]. The VWF: RCo and VWF: Ag levels were between 0.14 -0.33 U/dL and 0.08-0.25 iU/dL in pronounced VWD
type 1 and between 0.43-0.64 U/dL and 0.36-0.64 in LowVWF respectively. The PFA CT EPI and ADP were prolonged with values between 150-250 seconds in
6 of 10 and in 2 of 10 LowVWF patients. The PFA CT EPI and ADP were strongly prolonged above 250 seconds in VWD 2A, 2B, 2M and type 3. The PFA CT EPI
and ADP prologations in VWD type 1 are in between the values seen in LowVWF versus VWD type 2A, 2B and 2M.
References
- Michiels JJ, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, van de Velde A, et al. Characterization of recessive severe type 1 and 3 von Willebrand disease (VWD), asymptomatic heterozygous carriers versus blood group O-related von Willebrand factor deficiency and dominant type 1 VWD. Clin Apllied Thromb/Hemostas. 2006; 12: 277-295.
- Michiels JJ, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, van de Velde A, et al. Classification and characterization of hereditary types 2A, 2B, 2C, 2D, 2E, 2M, 2N, and 2U (unclassifiable) von Willebrand disease. Clin Apllied Thromb/Hemostas. 2006; 12: 397-420.
- Ruggeri ZM. Structure of von Willebrand factor and its function in platelet adhesion and thrombus formation. Best Practice Res Clin Haematol. 2001; 14: 257-279.
- Schneppenheim R, Budde U, Ruggeri ZM. A molecular approach to the classification of von Willebrand disease. Best Practice Res Clin Haematol. 2001; 14: 281-298.
- Hoyer LW, Rizza CR, Tuddenham D, Carta C, Armitage H, Rotblat F. Von Willebrand factor multimeric pattern in von Willebrand disease. Br J Haematol. 1983; 55: 493-507.
- Armitage H, Rizza CR. Two populations of factor VIII-related antigen in a family with von Willebrand’s disease. Br J Haematol. 1979; 41: 279-289.
- Ruggeri ZM, Pareti FI, Mannnucci PM, Ciavarella N, Zimmerman TS. Heightened interaction between platelet and FVIII/von Willebrand factor in a new subtype of von Willebrand disease. N Eng J Med. 1980; 302: 1047-1051.
- Zimmermann ThS, Dent JA, Ruggeri ZM, Nannini LH. Subunit composition of plasma von Willebrand factor; J Clin Invest. 1986; 77: 947-951.
- Schneppenheim R, Budde U. Phenotypic and genoypic diagnosis of von Willebrand disease. Sem Hematol. 2005; 42: 15-28.
- Sadler JE. A revised classification of von Willebrand disease. Throm Haemostas. 1994; 71: 251-279.
- Sadler JE, Mannucci PM, Bentorp E, Bochkov N, Boulyjenkov V, Ginsburg D, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemostas. 2000; 84: 160-174.
- Michiels JJ, van de Velden A, van Vliet HHDM, van der Planken M, Schroyens W, Berneman Z. Response of von Willebrand factor parameters to desmopressin in patients with type 1 and type 2 congenital von Willebrand disease: diagnostic and therapeutic implications. Sem Thromb Hemostas. 2002; 28: 111-131.
- Sadler JE, Budde U, Eikenboom, Favaloro EJ, Hill FGH, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemostas. 2006; 94: 2103-2114.
- Michiels JJ, van Vliet HHDM, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, et al. Intravenous DDAVP and factor VIII-von Willebrand factor concentrate for the treatment and prophylaxis of bleeding in patients with von Willebrand disease type 1, 2 and 3. Clin Applied Throm/hemostas. 2007; 13: 14-34.
- Fressinaud F, Veyradier A, Truchard F, Martin I, Boyer-Neumann C, Trossart M, et al. Screening for von Willebrand disease with a new analyzer using high shear stress: a study of 60 cases. Blood. 1998; 91: 1325-1331.
- Fressinaud F, Veyradier A, Signaud M, Boyer-Neumann C, LeBoterff C, Meyer D. Therapeutic monitoring of von Willebrand disease: interest and limits of a platelet function analyser at high shear rate. Br J Haematol. 1999; 106: 777-783.
- Mazurier C, Gaucher C, Jorieux S, Goydemand M. Biological effect of desmopressin in eight patients with type 2N (Normandy) von Willebrand disease. Br J Haematol. 1994; 88: 849-854.
- Caron C, Mazurier C, Goudemand J. Large experience with factor VIII binding assay of plasma von Willebrand factor using commercial reagents. Br J Haematol. 2002; 117: 716-718.
- Jacquemin M, Lavend’homme R, Benhida A, Vanzieleghem B, d’Oiron R, Lavergne JM, et al. A novel cause of mild/moderate Haemophilia A : mutations scattered in the factor VIII C1 domain reduce factor binding to von Willebrand factor. Blood. 2000; 96: 958-965.
- d’Orion R, Lavergne JM, Lanend’homme R, Benhida A, Jean-Claude Bordet, Negrier C, et al. Deletion of alanine in the FVIII C2 domain results in mild hemophilia by impairing FVIII binding to VWF and phospholipids. Blood. 2004; 103: 155-157.
- Schneppenheim R, Krey S, Bergman F, Bock D, Budde U, Lange M, et al. Genetic heterogeneity of severe von Willebrand disease type III in the German population. Hum genet. 1994; 94: 640-652.
- Zhang ZP, Blomback M, Egberg N, Falk G, Anvret M. Characterization of the von Willebrand factor gene in von Willebrand disease type III patients from 24 families of Swedish and Finnish origin. Genomics. 1994; 21: 189-193.
- Zhang A, lindstedt M, Blomback M. Effects of the mutant von Willebrand factor gene in von Willebrand disease. Hum Genet. 1995; 96: 388-394.
- Eikenboom JCJ Castaman G, Vos HL, Bertina RM, F Rodeghiero F. Characterization of genetic defects in recessive type 1 and type 3 von Willebrand patients of Italian origin. Thromb Haemostas. 1998; 79: 709-717.
- Baronciani L, Cozzi G, Cabciani MT, Peyandi F, Scrivastava A, Federici AB, et al. Molecular characterization of a multietnic group of 21 patients with type 3 von Willebrand disease. Thromb Haemostas. 2000; 84: 536-540.
- Eikenboom JCJ. Congenital von Willebrand disease type 3: clinical manifestations, pathophysiology and molecular biology. Best Practice Res Clin Haematol. 2001; 14: 365-379.
- ISTH data base of von Willebrand factor mutations: www.shef.ac.uk/vwf
- Allen S, Abuzenadah AM, Hinks J, Blagg JL, Gursel T, Ingerslev J, et al. A novel von Willebrand disease-causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerisation and secretion. Blood. 2000; 96: 560-568.
- Castaman G, Lattuada A, Mannucci PM, Rodeghiero F. Factor VIII: C increases after desmopressin in a subgroup of patients with autosomal recessive severe von Willebrand disease. Brit J Haematol. 1995; 89: 147-151.
- Castaman G, Eikenboom JCJ, Lattuada A, Mannucci PM, Rodeghiero F. Heightened proteolysis of the von Willebrand factor subunit in patients with von Willebrand disease hemizygous or homozygous for the C2364F mutation. Brit J Haematol. 2000; 108: 188-190.
- Castaman G, Novella E, Castiglia E, Eikenboom JCJ, Rodeghiero F. A novel family with recessive von Willebrand disease due to compound heterozygosity for a splice site mutation and a missense mutation in the von Willebrand factor gene. Thromb Res. 2002; 105: 135-138.
- Tjernberg P, Castaman G, Vos HL, Bertina RM, Eikenboom JCJ. Homozygous C2362F von Willebrand fiactor induces intracellular retention of mutant von Willebrand factor in autosomal recessive severe von Willebrand disease. Br J Haematol. 2006; 133: 409-418.
- Castaman G, Bertoncello, Bernardi M. Eikenboom JC, Budde U, Rodeghiero F. Autosomal recessive von Willebrand disease associated with compound heterozygosity for a novel nonsense mutation (2908delC) and the missense mutatio C2362F: definite evidence for non-penetrance of the C2363F mutation. Am J Hematol. 2007; 82: 376-380.
- Schneppenheim R, Budde U, Obser T, Brassard J, Mainush K, Ruggeri ZM, et al. Expression and characterization of von Willebrand factor dimerization defects in different types of von Willebrand disease. Blood. 2001; 97: 2059- 2066.
- Ruggeri ZA, Nilsson IM, Lombardi R, Holmberg L, Zimmerman ThS. Aberrant multimeric structure of von Willebrand factor in a new variant of von Willebrand’s disease (type IIC). J Clin Invest. 1982; 70: 1124-1127.
- Holmberg L, Karpman D, Isakson C, Kristofferson AC, Lethagen S, Schneppenheim R. Ins405AsnPro mutation in the von Willebrand factor propeptide in recessive type 2A (IIC) von Willebrand’s disease. Thromb Haemostas. 1998; 79: 718-722.
- Mazurier C, Manucci PM, Parquet-Gernez A, Goudemand, Meyer D. Investigation of a case of subtype IIC von Willebrand disease: characterisation of the variability of this subtype. Amer J Hematol. 1986; 22: 301-311.
- Gaucher C, Diéval J, Mazurier C. Characterization of von Willebrand factor gene defects in two unrelated patients with type IIC von Willebrand disease. Blood. 1994; 84: 1024-1030.
- Batlle J, Lopez-Fernandez MF, Lasiera J, Fernandez Villamor AF, Lopez Berges C, Lopez Borrasca A, et al. Von Willebrand disease type IIC with different abnormalities of von Willebrand factor in the same sibship. Amer J Hematol. 1986; 21: 177-188.
- Batlle J, Lopez Gernandez MF, FernandezVillamor A, Lopez Berges C, Zimmerman ThS. Multimeric pattern discrepancy between platelet and plasma von Willebrand factor in type IIC von Willebrand disease. Amer J Hematol. 1986; 22: 87-88.
- Schneppenheim R, Thomas KB, Krey S, Budde U, Jessat U, Sutor AH, et al. Indentification of a candidate missense mutation in a family with von Willebrand disease type IIC. Hum Genet. 1995; 95: 681-686.
- Casana P, Martinez F, Haya S, Espinos C, Aznar JA. Association of the T1156M mutation in the von Willebrand factor gene with dominant type 1 von Willebrand disease. Ann Hematol. 2001; 80: 381-383.
- Lethagen S, Isaksson C, Schaedel C, Holmberg L. Von Willebrand disease caused by compound heterozygosity for a substitution mutation (T1156M) in the D3 domain of the von Willebrand factor and a stop mutation (Q2470X). Thromb Haemostas. 2002; 88: 421-426.
- Eikenboom JCJ, Matsushita T, Reitsma PH, Tuley E, Castaman G, Briët E, et al. Dominant type 1 von Willebrand disease caused by mutated cysteine residues in the D3 domain of von Willebrand factor. Blood. 1996; 88: 2433-2441.
- Castaman G, Eikenboom JCJ, Missiaglia E, Rodeghiero F. Autosomal dominant type 1 von Willebrand disease due to C1130F mutation in exon 26 of von Willebrand factor gene: description of five families and evidence for a founder effect. Br J Haematol. 2000; 108: 876-879.
- Tjernberg P, Vos HL, Castaman G, Bertina RM, Eikenboom JCJ. Dimerization and multimerization defects of von Willebrand factor due to mutated cysteine residues. J Thromb Haemostas. 2004; 2: 257-265.
- Haberichter SL, balistrreri M, Christopherson P, Morateck P, Gavazova S, Bellissimo DB, et al. Assay of von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006; 108: 3344-3351.
- James PD, O’Brien LA, Hegadorn CA, Notley CRP, Sinclair GD, Huogh C, et al. A novel type 2A von Willebrand factor mutation located at the last nucleotide of exon 26 (3538G>A) causes skipping of 2 non-adjecent exons. Blood. 2004; 104: 2739-2745.
- Schneppenheim R, Marggraf O, Eckert K, Obser T, Oyen F, Pieconka A, et al. Molecular background of smeary’ von Willebrand factor multimers. Annual Meeting American Society of Hematology. Blood. 2007; 110: 709.
- Schneppenheim R, Lenk H, Obser T, Oldenburg J, Oyen F, Schneppenheim S, et al. Recombinant expression of mutations causing von Willebrand disease type Normandy: characterization of a combined defect of factor VIII binding defect and multimerization. Thromb Haemostas. 2004; 92: 36-41.
- Casonato A, Sartorello F, Cattini MG, Pontera E, Soldera C, Bertomoro A, et al. An Arg760Cys mutation in the consensus sequence of the von Willebrand factor propeptide cleavage site is responsible for a new von Willebrand disase variant. Blood. 2003; 101: 151-156.
- Hilbert L, Nurden P, Caron C, Nurden AT, Goudemand J, Myer D, et al. Type 2N von Willebrand disease due to compound heterozygosity for R854Q and a novel R763G mutation at the cleavage site of von Willebrand factor propeptide. Thromb Haemostas. 2006; 96: 290-294.
- Goodeve A, Eikenboom JCJ, Castaman G, Rodeghiero F, Federici AB, Batlle J, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of type 1 von Willebrand Disease (MCMDM-1VWD). Blood. 2007; 109: 112-121.
- Castaman G, Lethagen S, Federici AB, tosetto A, Goodeve A, Budde U, et al. Response to desmopressine is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European study MCMDM-1VWD. Blood. 2008: 111: 3531-3539.
- Budde U, Schneppenheim R. Eikenboom JCJ, Goodeve A, Will K, Drewke E, et al. Detailed von Willebrand factor multimer analysis in patients with von Willebrand disease in the uropean study, Molecular and Clinical Markers for the Diagnosis and Management of type 1 von Willebrand disease (MCMDM- 1VWD). J Thromb Haemostas. 2008; 6: 762-771.
- James PD, Noley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007; 109: 145-154.
- Cumming A, Grundy P, Keeney S, Lester W, Enayat S, Guillat A, et al. An investigation of von Willebrand factor genotype in UK patients diagnosed to have type 1 von Willebrand disease. Thromb Haemostas. 2006; 96: 630-641.
- Collins PW, Cumming AM, Goodeve A, Lillicrap D. Type 1 von Willrand disease : applicsation of emerging data to clinical practice. Haemophila. 2008; 14: 685-696.
- Michiels JJ, Berneman Z, Gadisseur A, Van Der Planken M, Schroyens W, Van Vliet H. Laboratory diagnosis and molecular basis of mild von Willebrand disease type 1. Acta Haematol. 2009; 121: 89-97.
- De Wit TR, Van Mourik JA. Biosynthesis, processing and secretion of von Willebrand factor: biological implications. Best Prractice ResClin Haematol. 2001; 14: 241-255.
- Eikenboom JCJ, Castaman G, Kamphuizen PW, Rosendaal FR, Bertina RM. The factor VIII/von Willebrand factor ratio discriminates between reuced synthesis and increased clearance of von Willebrand factor. Thromb Haemostas. 2002; 87: 252-257.
- Huang RH, Wang Y, Roth R, Yu X, Purvis AR, Heuser JE, et al. Assembly of Weibel-Pallade bodies-like tubules from N-terminal domains of von Willebrand factor. Proc Nat Acad Sci USA. 2008;105: 482-487.
- Haberichter SL, Castaman G, Budde U, Peak I, Goodeve A, Rodeghiero F, et al. Identification of type 1 von Willebrand disease with reduced von Willebrand factor survival by assay of VWF propeptide in the European MCMDM-1VWD study. Blood. 2008; 111: 4979-4985.
- Davies JA, Collins PW, Hathaway IS, Bowen DJ. Von Willebrand factor: evidence for variable clearance in vivi according to Y/C1584 phenotype and ABO blood group. J Thromb Haemostas. 2008; 6: 97-103.
- Gadisseur A, Berneman Z, Schroyens W, Michiels JJ. Laboratory diagnosis of von Willebrand disease type 1/2E (2A subtype IIE) type 1 Vicenza and mild type 1 caused by mutations in the D3, D4, B1-B3, and C1-C2 domains of the von Willebrand factor gene. Acta Haematol. 2009; 121: 128-138.
- Szukowska M, Gallinaro L, Cattini MG, Pontara E, Sartorello F Daldone V, Prandoni P, et al. von Willebrand factor propeptide makes it easy to identify the shorter von Willebrand factor survival in patients with type 1 Vicenza von Willebrand disease. Brit J Haematol. 2009; 143: 107-114.
- Smejkal P, Michiels JJ, Zapletal O, Blatny J, Batorova A, Pricangova T, et al. European Clinical, Laboratory and Molecular (2020 ECLM) Diagnostic Work-up and Classification of von Willebrand Disease from the perspectives of clinicians and Scientists. Thromb Haemstas Res. 2019: 3: 1032.
- Michiels JJ, Smejkal P, Mayger K, Moore G, Budde U, Berneman Z, et al. Combined use of rapid von Willebrand fctor (VWF) activity, VWF-propeptide and classical VWF assays for improved diagnosis of von Willebrand disease type 1, 2N, and 2E due to mutsations in the D1, D2, D’, D3 and D4 domains of the VWF gene. Thromb Haemostas Res. 2019; 3: 1027.
- Michiels JJ, Smejkal P, Mayger K, Moore G, Blatny J, Penka M, et al. Superiority of te rapid von Willebranfd factor (VWF) VWF: GPIbR and VWF: GPIbM assays in type 2A, 2B and 2M von Willebrand disease. Int J Clin Exp Med Sci. 2019; 5: 80-91.
- Michiels JJ, Smejkal P, Penka M, Blatny J, Budde U, Hermans C, et al. Basic insights into the molecular etiology and pathology of von Willebrand factor (VWF) behind the ISTH and ECLM classifications of von Willebrand disease using a complete set of VWF parameters. Austin Hematol. 2019; 4: 1026.
- Nummi V, Lassila R, Joutsi-Korhonen L, Armstrong E, Szanto T. Comprehensive re-evaluation of historical von Willebrand disease diagnosis in association with whole blood platelet aggregation and function. Int J Lab Hem. 2018; 40: 304-311.
Citation: Michiels JJ, Smejkal P, Penka M, Budde U, Hermans C, Blatny J, et al. Comparative Analysis of the European, Canadian and UK ISTH Defined VWD Type 1 Prospective Studies: Principles to Translate the ISTH into the ECLM Classification of Von Willebrand Disease on Top of Sensitive Von Willebrand Factor Assays and Multimeric Analysis in Medium to High SDS Resolution Gels. Thromb Haemost Res. 2020; 4(3): 1049.