
Case Report
Thromb Haemost Res. 2021; 5(2): 1058.
Acquired Hemophilia a Secondary to Autoimmune Pancreatitis with Elevated IG4
Li Y¹, Li J¹, He H², Cai R¹ and Feng Q¹*
1Department of Clinical Laboratory, Tai’an Central Hospital, Shandong Province, China
2Department of Blood Transfusion, Tai’an Hospital of Traditional Chinese Medicine, Shandong Province, China
*Corresponding author: Qiang Feng, Department of Clinical Laboratory, Tai’an Central Hospital, 29th Longtan Road, Tai’an City, Shandong Province, 271000, China
Received: March 22, 2021; Accepted: April 16, 2021; Published: April 23, 2021
Abstract
Acquired Hemophilia A (AHA) is a rare disease resulting from autoantibodies against Factor VIII (FVIII) that leads to bleeding. AHA associated with IgG4 related diseases is even rarer. The patient was diagnosed with IgG4 associated autoimmune pancreatitis in January 2019, and the condition improved after two hospitalizations. However, 22 months later, the patient was admitted to hospital due to generalized bleeding points. He was diagnosed with AHA and improved after hormone therapy and plasma exchange. Although IgG4 is associated with IgG4-related disease and AHA, its relevance to the etiology of both diseases is not well understood.
Introduction
Autoimmune pancreatitis (autoimmune pancreatitis, AIP) is a special type of chronic pancreatitis mediated by autoimmunity [1]. Type 1 AIP is local manifestations of IgG4 related diseases (IgG4 related disease, IgG4 RD) in pancreas which is often accompanied by extrapancreatic manifestations, such as sclerosing cholangitis, sialitis, dacryitis, retroperitoneal fibrosis, lymphadenopathy, interstitial pneumonia and renal tubulointerstitial nephritis. Glucocorticoid is effective, but often relapses [2].
Acquired Hemophilia A (AHA) is a disease caused by acquired autoantibodies that inhibit Factor VIII (FVIII) activity, and it leads to severe hemorrhages. The antibodies are mostly sub-classified as IgG4 and IgG1 [3]. Acquired hemophilia A associated with IgG4 related diseases is rare.
Case Presentation
A 65-year-old man was admitted to the stomatology department of our hospital due to bilateral submandibular gland tumors in November 2018. His laboratory findings showed raised serum IgG concentration of 44.34 g/l and slightly prolonged plasma Prothrombin Time (PT) 14.30s, while the other laboratory results were basically normal. The patient was discharged three days later without further diagnosis and treatment.
Due to skin and sclera yellow staining, skin pruritus, white pottery soil and weight loss of 5kg, the patient underwent MRI examination in a lower hospital, which indicated the tumor of common hepatic duct, so he was transferred to our hospital and admitted to the oncology department in January 2019. His laboratory findings showed raised serum IgG concentration of 44.34 g/l, hepatobiliary enzymes increased significantly and tumor markers elevated as shown in Table 1. Computed tomography showed Intrahepatic bile duct dilatation, gallbladder atresia, annular thickening of middle and upper segment of common bile duct, mild dilatation of bile duct in left lobe of liver, abnormal enhancement of pancreas and kidney. Comprehensive consideration was that sclerosing cholangitis and autoimmune pancreatitis were more likely. He was treated with Hydroprednisone (30mg/day) and Polyene phosphatidylcholine (465mg/day) for 3 days. The patient’s condition improved and asked to be discharged.
![]()
2018.11
2019.01
2019.05
2020.10
Complete blood counts
Wbc
-
4.22*109/L
4.66*109/L
4.26*109/L
RBC
-
3.58*1012/L
3.55*1012/L
3.01*1012/L
Hb
-
111g/l
112g/l
92g/l
PLT
-
239*109/L
209*109/L
203*109/L
Coagulation
PT
14.3s
14.2s
14.7s
12.5s
APTT
23.5s
20.5s
31.2s
63.6s
Fib
3.77g/l
3.37g/l
3.92g/l
5.43g/l
FDP
-
-
2mg/l
9.5mg/l
D-Dimer
1.03mg/l
0.73mg/l
0.63mg/l
3.88mg/l
Table 1: Laboratory examination results of four hospitalizations.
In May 2019, the patient was re-admitted to the hospital for hormone therapy, and laboratory tests showed that the increased IgG4 (2820mg/dl, normal range: 4.8-105 mg/dl) supported IgG4-related diseases. After hormone treatment, the CT results showed that the wall of the middle and upper segment of common bile duct was slightly thicker and lighter than before; the dilatation of bile duct in the left lobe of the original liver was not obvious; the abnormal enhancement area of pancreas was reduced; and the abnormal enhancement area of both kidneys was similar. The elevations of hepatobiliary enzymes and tumor markers improved. After discharge, oral administration of prednisone and mycophenolate mofetil dispersible tablets was continued.
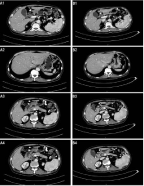
Figure 1: Images of pancreas and gallbladder. A1, Intrahepatic bile duct
dilatation, gallbladder atresia, annular thickening of middle and upper
segment of common bile duct. B1, the wall of the middle and upper segment
of common bile duct was slightly thicker and lighter than before. A2, mild
dilatation of bile duct in left lobe of live. B2, the dilatation of bile duct in the
left lobe of the original liver was not obvious. A3, the density of uncinate
process decreased significantly. B3, the density of uncinate process was not
significantly decreased. A4, abnormal enhancement of pancreas. B4, the
abnormal enhancement area of pancreas was reduced.
In October 2020, the patient was admitted to the emergency intensive care unit of our hospital because of “extensive bleeding point in the whole body for one week, aggravation and epistaxis for one day”. Bleeding could be seen in the oral mucosa, multiple bleeding points in the forearm and abdomen, and ecchymosis could be seen in the foot. Blood cell counts showed anemia, and the serum IgG4 was 4340 mg/dl. Antinuclear antibody was negative. Anti-SS-B and SS-A antibodies were negative. APTT was prolonged to 63.6s whereas the platelet count and prothrombin time were within normal ranges. Fibrin degeneration product and d-dimer were elevated. APTT immediate correction test was negative, APTT prolonged incubation test was positive, suggesting the presence of time-dependent clotting inhibitors. FVIII activity was 1.2%, and FVIII inhibitor was elevated to 66.56 BU/ml. Based on the clinical findings and laboratory data, he was diagnosed with AHA. After hormone therapy and plasma exchange, the patient had no active bleeding and was discharged with FVIII activity was elevated to 3.2% and FVIII inhibitor dropped to 33.2 BU/ml although APTT was still prolonged as shown in Table 1.
Discussion
IgG4-related disease is a chronic progressive autoimmunerelated disease, which is mainly characterized by increased IGG4, IGG4 positive plasma cell infiltration and fibrosis in the affected tissues. The clinical symptoms of the patients vary from organ to organ. The early stage of the disease is often misdiagnosed and missed because of the non-specific clinical manifestations. The disease can involve any organ and often involves multiple organs at the same time. IgG4-associated autoimmune pancreatitis is often characterized by obstructive jaundice caused by organ swelling and pancreatic duct compression. IgG-RD has a variety of clinical manifestations and pathological changes, and there is still a lack of gold standard for diagnosis. IgG-RD crosses and overlaps with many diseases in clinical, pathological and laboratory examination, but most of them respond well to hormones, so early correct diagnosis is very important.
In this present case, the serological examination, CT examination and clinical manifestations of the patient were longitudinally analyzed as follows: 1. The serum globulin, IGG and IG4 of the patient always increased, and the IGG4 decreased from 2820mg/dl to 843mg/dl after hormone therapy. 2. The results of CT before hormone treatment were consistent with sclerosing cholangitis and autoimmune pancreatitis, and were significantly improved after hormone treatment. 3. The patients had submandibular gland mass, painless jaundice, fatigue and weight loss successively, and the symptoms were significantly improved after hormone treatment. It has been reported that when the cut-off value of serum IgG4 is 135mg/dl, the sensitivity of diagnosing AIP is 65% and the specificity is 98% [4]. A meta-analysis including 15 original studies also found that serum IgG4 had lower sensitivity and higher specificity for the diagnosis of AIP [5]. In this case, no biopsy and pathological examination were performed before and after four hospitalizations. However, comprehensive consideration suggested that the possibility of IgG4 related diseases was high.
Acquired Hemophilia A (AHA) is a rare disease resulting from autoantibodies against FVIII that leads to bleeding, which is often spontaneous and severe. This results in a bleeding diathesis in patients who previously had no personal or family history of bleeding [6]. Almost half of the cases are idiopathic. The others are related to autoimmune diseases, malignancies, pregnancy, infections or medications [6-7]. Systemic Lupus Erythematosus (SLE) is the most common autoimmune disease associated with AHA, and AHA secondary to IGG4-RD is very rare. Sugino et al. and Narazaki et al. reported the only two cases of AHA associated with IgG4-related AIP [8-9]. In addition, two cases of AHA associated with elevated IgG4 were also reported [10-11].
The main IgG subclasses of FVIII-binding antibodies in healthy per- sons were IgG1 and IgG3, and their titers were low. In contrast, IgG1 and IgG4 antibodies were most prominent in patients with AHA, and the titers of FVIII-binding antibodies were well correlated with inhibitor titers [12]. In the case reported by Narazaki et al. even after the clinical symptoms of AIP improved, the serum IgG4 level remained high until the treatment of AHA was started [9]. However, in this present case, the patient developed AHA after the obvious improvement of AIP symptoms, although the level of serum IGG4 was still high, it was significantly lower than that before hormone therapy. Nagao et al. also reported serum IgG4 was not elevated when AHA relapsed [10]. Although IgG4 is associated with both diseases, its relevance to the etiology of IgG4-related disease and AHA is not well understood.
In conclusion, IGG4-related diseases complicated with AHA are very rare. Although some scholars believe that AHA can be caused by IgG4-related diseases, the immune relationship between IGG4 and AHA could not be determined. In the future, it is important to focus on the role of IgG4 antibodies in order to understand the clinicopathological characteristics of IgG4-related diseases and AHA.
Statement of Ethics
The study was approved by the Ethics Committee of Tai’an Central Hospital and the informed consent of the patients was obtained.
Author Contributions
Yi Li, Jianmin Li, Hualin He, Ruimin Cai and Qiang Feng have (1) made substantial contributions to the conception or design of the work, or to the acquisition, analysis, or interpretation of data for the work; (2) participated in drafting the work or revising it critically for important intellectual content; (3) approved the final version to be published; and (4) agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
References
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001; 344: 732-738.
- Wu W, Yao X, Lin C, Jin D, Wang D, Lou W, et al. Should Steroid Therapy Be Necessarily Needed for Autoimmune Pancreatitis Patients with Lesion Resected due to Misdiagnosed or Suspected Malignancy? Gastroenterol Res Pract. 2014; 2014: 253471.
- Fulcher CA, de Graaf Mahoney S, Zimmerman TS. FVIII inhibitor IgG subclass and FVIII polypeptide specificity determined by immunoblotting. Blood. 1987; 69: 1475-1480.
- van Heerde MJ, Buijs J, Hansen BE, de Waart M, van Eijck CH, Kazemier G, et al. Serum level of Ca 19-9 increases ability of IgG4 test to distinguish patients with autoimmune pancreatitis from those with pancreatic carcinoma. Dig Dis Sci. 2014; 59: 1322-1329.
- Lian MJ, Liu S, Wu GY, Liu SY. Serum IgG4 and IgG for the diagnosis of autoimmune pancreatitis: A systematic review with meta-analysis. Clin Res Hepatol Gastroenterol. 2016; 40: 99-109.
- Franchini M, Vaglio S, Marano G, Mengoli C, Gentili S, Pupella S, et al. Acquired hemophilia A: a review of recent data and new therapeutic options. Hematology. 2017; 22: 514-520.
- Collins P, Baudo F, Huth-Kühne A, Ingerslev J, Kessler CM, Castellano ME, et al. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Res Notes. 2010; 3: 161.
- Sugino K, Gocho K, Ishida F, Kikuchi N, Hirota N, Sato K, et al. Acquired hemophilia A associated with IgG4-related lung disease in a patient with autoimmune pancreatitis. Intern Med. 2012; 51: 3151-3154.
- Narazaki T, Haji S, Nakashima Y, Tsukamoto Y, Tsuda M, Takamatsu A, et al. Acquired hemophilia A associated with autoimmune pancreatitis with serum IgG4 elevation. Int J Hematol. 2018; 108: 335-338.
- Nagao Y, Yamanaka H, Harada H. A patient with hypereosinophilic syndrome that manifested with acquired hemophilia and elevated IgG4: a case report. J Med Case Rep. 2012; 6: 63.
- Li X, Duan W, Zhu X, Xu J. Immunoglobulin G4-related acquired hemophilia: A case report. Exp Ther Med. 2016; 12: 3988-3992.
- Hofbauer CJ, Whelan SF, Hirschler M, Allacher P, Horling FM, Lawo JP, et al. Affinity of FVIII-specific antibodies reveals major differences between neutralizing and nonneutralizing antibodies in humans. Blood. 2015; 125: 1180-1188.