
Special Article – Anticoagulants
Thromb Haemost Res. 2021; 5(2): 1060.
Effects of Long-term Direct Thrombin Inhibition by Dabigatran Etexilate on Progression of Atherosclerosis in ApoE-/- and LDLR-/- Double-knockout Mice
Hyodo K¹, Sanda T¹, Yoshimura M¹, Ishii H² and Yamashita T¹*
¹Laboratory of Medical Technology, Faculty of Nutrition, Kobe Gakuin University, Kobe, Japan
²Medical Corporation, Jinkeikai Ishii Hospital, Akashi, Japan
*Corresponding author: Tsutomu Yamashita, Laboratory of Medical Technology, Faculty of Nutrition, Kobe Gakuin University, 518, Arise, Igawadani-Cho, Nishiku 651-2180, Kobe, Japan
Received: March 29, 2021; Accepted: April 28, 2021; Published: May 05, 2021
Abstract
Background: Atherosclerosis is characterized by a hypercoagulable state, during which coagulation and fibrinolytic factors are simultaneously activated. However, details regarding the progression of atherosclerosis remain unknown. Here, we investigated the effects of direct long-term inhibition of thrombin by dabigatran etexilate on atherosclerotic progression in apolipoprotein E–/– and low-density lipoprotein receptor–/– double-knockout mice.
Methods: Mice received either standard chow (placebo group) or dabigatran-supplemented chow for 22 weeks. The amount of atherosclerosis was estimated as the ratio of the atherosclerotic area to the total aortic intimal area. Immunohistochemistry was used to examine the expression of Matrix Metalloproteinase-9 (MMP-9), Vascular Endothelial Growth Factor (VEGF), Tissue-Type Plasminogen Activator (t-PA), and Endothelial Nitric Oxide Synthase (eNOS) in atherosclerotic regions.
Results: The atherosclerotic area was smaller in the dabigatran group than in the placebo group. Immunohistochemistry revealed decreased expression of MMP-9 and VEGF, but increased expression of eNOS, in the dabigatran group compared with the placebo group. t-PA expression did not differ between the groups.
Conclusion: Direct long-term inhibition of thrombin by dabigatran in mice led to a decrease in atherosclerosis progression via decreased expression of MMP-9 and VEGF.
Keywords: Atherosclerosis; Dabigatran etexilate; MMP-9; Thrombin; VEGF
Introduction
Atherosclerosis is an inflammatory disease in which the arteries gradually narrow and stiffen over time. Although it is a leading cause of acute coronary syndrome and cardiovascular disease in humans, the precise mechanisms underlying its development remain unknown [1]. Clinically, atherosclerosis is characterized by the development of atherosclerotic plaque within the tunica intima, the innermost layer of the artery wall. Over time, the plaque enlarges until the endothelium separating the plaque from the blood circulation ruptures, causing circulating platelets to be exposed to the thrombogenic cellular and acellular components of the plaque. This exposure initiates the coagulation cascade leading to thrombus formation and possible luminal occlusion [2].
Thrombin is a serine protease that is best known for its crucial role in feedback activation of the coagulation cascade. This role obviously makes thrombin an important enzyme after rupture of an atherosclerotic plaque. However, thrombin is also a multifunctional protein that plays important roles outside the coagulation cascade. For example, thrombin increases endothelium-dependent vasorelaxation [3], increases vascular inflammation [4], and alters endothelial cell phenotype [5]. Together, these effects likely decrease endothelial function and induce vascular smooth muscle cell contraction and proliferation-changes known to accelerate atherogenesis. During atherogenesis, macrophages localize at sites of inflammation and release tissue factor for the initiation of the thrombin formation [6]. Thrombin also accelerates T-cell proliferative responses, including cytokine production, induction of leukocyte chemotaxis, and immunoresponsiveness [7,8]. Together, these findings suggest that thrombin and the coagulation cascade are involved not only in plaque rupture but also in the development and progression of atherosclerosis and plaque formation; however, the underlying mechanisms remain to be elucidated.
Many oral anticoagulants are currently approved for the treatment of nonvalvular atrial fibrillation [9]. Therefore, it is very interesting whether the inhibition of the thrombin decreases the progression of atherosclerosis. Dabigatran etexilate is an oral anticoagulant developed specifically for long-term administration [10]. It is currently used for the treatment of nonvalvular atrial fibrillation, but its clinical application is extending with its application as an inhibitor of activated coagulation factor X in second-generation anticoagulant therapy to replace warfarin [11]. Dabigatran may also reduce the risk of bleeding during anticoagulation therapy [9].
Here, we examined the effects of long-term (22-week) thrombin inhibition by dabigatran on the development and progression of atherosclerosis in Apolipoprotein E (ApoE)–/– and Low-Density Lipoprotein Receptor (LDLR)–/– double-knockout mice. Histological and immunological examinations of the whole aorta allowed us to compare the sites of plaque development as well as the progression of atherosclerosis in mice with or without thrombin inhibition.
Materials and Methods
Experimental animals and drug administration
C57BL/6 mice (age, 10-13 weeks) were obtained from SLC (Hamamatsu, Japan). Double- homozygous Apo-E-/- and LDLR-/-- deficient mice (129 × C57BL/6J background) were obtained from The Jackson Laboratory (Bar Harbor, Maine, USA) and bred through brother-sister mating. All animals were maintained at Kobe Gakuin University (Hyogo, Japan) in air- conditioned rooms (22.5 ± 0.5°C; humidity, 50% ± 5%) on a 12:12-h light:dark photocycle with free access to chow and drinking water. Only male mice were used in the experiments. All procedures were conducted in compliance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Science of the Physiological Society of Japan.
Three groups of animals were used. One control group was used in which C57BL/6 mice were fed placebo chow. In addition, two experimental groups were used in which Apo-E-/- and LDLR-/- double-knockout mice (n=38 overall) were fed placebo chow or dabigatran- supplemented chow. In the three groups, 6-week-old mice were given either placebo chow or dabigatran-supplemented chow (BIBR1048MS; 7.5mg dabigatran/g) for 22 weeks; at 22 weeks, the mice were examined. Placebo chow and dabigatran-supplemented chow (0mg dabigatran/g-chow, 7.5mg dabigatran/g-chow) were provided by the manufacturer (Boehringer Ingelheim, Ingelheim am Rhein, Germany) and had been adjusted on the basis of a highfat diet (ER R/M acc D122451(II)mod; ssniff, Soest, Germany). The dose of dabigatran reflects the low bioavailability of dabigatran in mice (about 6.5%), thus necessitating a relatively high dose for oral administration [12,13].
Plasma concentration and efficacy of dabigatran
After the mice had been fed for 22 weeks, blood was collected from the abdominal aorta of each mouse into 3.14% sodium citrate via a 23-gauge needle and stored at -80°C until analysis.
Plasma concentrations of dabigatran were measured by using the Hemoclot Thrombin Inhibitors assay (Hyphen Biomed, Neuvillesur- Oise, France) in accordance with the manufacturer’s instructions but with slight modifications. In brief, mouse plasma was diluted 1:8 with 0.9% NaCl solution; 37.5μL of this dilution was mixed with 75μL of human plasma. After incubation of the mixture for 1min at 37°C, 75μL of thrombin solution was added and the clotting time was measured by using a coagulometer (CA-101, Sysmex, Kobe, Japan). For quantification purposes, dilutions of Dabigatran Calibrator Low (Hyphen Biomed) covering the range of 0-500ng/mL were run in parallel. All experiments were performed in duplicate.
Bleeding time
Mice were anesthetized with sodium pentobarbital (60mg/kg, intraperitoneal injection) and bleeding time was determined. In brief, a 3mm tail-tip transection was made, and blood drops were removed every 15s by using filter paper. If blood flow did not reoccur within 30s of wiping away a blood drop, bleeding was considered stopped. Experiments were terminated after 30min if the blood flow had not ceased.
Assessment of atherosclerosis progression
Atherosclerosis progression was assessed by estimating the area of an atherosclerotic region as a percentage of the entire surface area of the aorta, as previously described [14]. In brief, hearts were exposed through abdominal incision, and phosphate-buffered saline (PBS; pH 7.4) followed by 10% neutral-buffered formalin solution (Nacalai Tesque, Tokyo, Japan) was infused through an indwelling 20-gauge butterfly needle (Top Kasei, Tokyo, Japan). Next, the major blood vessels were washed with PBS and fixed with 10% neutralbuffered formalin solution by reflux through a femoral artery. Then, connective tissue and minor branching blood vessels were removed from the aortic arch. The extracted vessels were kept in 10% neutralbuffered formalin solution until processing, at which point they were incised along the longitudinal plane and pinned flat. The tissues were washed with distilled water for 30s, treated with 60% isopropyl alcohol for 1min, and stained with Oil Red O (to identify atherosclerotic plaques) at 37°C for 15min. Finally, the tissues were washed with 60% isopropyl alcohol and distilled water.
Stained specimens were photographed (Pentax K-7, Ricoh, Tokyo, Japan) and the images were transferred to a personal computer and analyzed with image analysis software (Image- Pro Plus, Media Cybernetics, Rockville, Maryland, USA). The whole area (W) of the dissected aorta and the portion that was stained positively with Oil Red O (R) were calculated; the resulting ratio ((R ÷ W) × 100%) was used as an index of atherosclerotic progression.
Histological and immunological analyses
The aortic root, aortic arch, and brachiocephalic artery were all analyzed by immunohistochemistry and histology; however, only data from the aortic arch are shown. The blood vessels of interest were fixed in OCT Compound (Tissue-Tek, Sakura, Japan) and frozen by using a dry ice - acetone mixture or liquid nitrogen. Frozen OCT-embedded tissue blocks were cut into 6μm sections, which were placed on poly-L-lysine-coated microscope slides (Muto Pure Chemicals, Tokyo, Japan).
For histological analysis, sections were stained with hematoxylin and eosin followed by elastica van Gieson stain (Merck KGaA, Darmstadt, Germany). Elastica van Gieson staining was performed according to a standard protocol [15]. For visualization of the elastin laminae and its fragments, the sections were stained by using an elastica van Gieson staining kit. The presence of calcium deposits was assessed by means of von Kossa staining in accordance with the supplier’s protocols (Genmed, USA). The specimen was positioned under an ultraviolet light for 1h in the dark, rinsed 3 times with double-distilled water, and treated with 5% sodium thiosulfate to remove background staining. After three washes with PBS, the specimen was counterstained with Nuclear Fast Red Solution for 5min.
For immunohistochemical analysis, the heart was exposed, and a butterfly catheter was inserted into the left ventricle. The heart was flushed with 10mmol/L PBS (pH 7.4) for about 3min to remove all blood. The blood vessels then were perfusion-fixed with 4% paraformaldehyde in PBS (Wako Pure Chemical Industries, Osaka, Japan).
For immunohistochemical analysis, after immunoperoxidase staining, sections were examined with anti-MMP-9 antibody (dilution, 1:100; MMP-9 Rabbit PAb, Bio Vision, California, USA), anti-VEGF antibody (1:100; VEGF Rabbit PAb, GeneTex, Irvine, California, USA), anti-t-PA antibody (1:100; t-PA Rabbit PAb, LabVision), and anti-Endothelial Nitric Oxide Synthase (eNOS) antibody (1:100; eNOS Rabbit PAb, LabVision). Epitopes recognized by the primary antibody were visualized by labeling with streptavidin and biotinylated horseradish peroxidase (LSAB2 Kit, Dako, Kyoto, Japan), as described previously. The slides then were counterstained with Mayer’s hematoxylin (TA-125-MH, LabVision) in accordance with standard protocols. During histology, cross-sections were obtained from at least three sections (luminal side, intraplaque, and intimal side) of each plaque.
For evaluation of the relative intensities of the stained areas, staining intensity was quantified by using digital image analysis software (Image Pro Plus Version; Media Cybernetics) followed by application of an immunochemistry data analysis protocol. The staining intensity was determined by means of a modified reciprocal intensity method. The reciprocal intensity was determined in a constant area that contained atherosclerotic plaque per section and then the results per mouse were averaged.
Statistical analysis
Results are expressed as means ± SEM. Comparisons among groups were made by using factorial ANOVA. Differences between groups of each determined parameter were analyzed by using Student’s t-test. A P value of <0.05 was defined as statistically significant. No adjustments were made. All analyses were accomplished by using the statistical package JMP (JMP 13 SAS Institute, Tokyo, Japan).
Results
Body weight and food intake
At 22 weeks, control mice weighed 36.7 ± 1.0 g (mean ± SEM; n=14), placebo mice weighed 49.5 ± 1.3 g (n=32), and dabigatran mice weighed 48.0 ± 1.4 g (n=23). Body weight did not differ significantly between the placebo and dabigatran mice, both of which were on a high-fat diet. Feed intake did not differ between the control and dabigatran mice.
Plasma concentration of dabigatran
In the mice fed the supplemented diet, the dabigatran plasma concentration was 64.5 ± 8.1 ng/mL (n=9) (data not shown). The anticoagulant efficacy of dabigatran was examined by deriving the international normalized ratio from the thrombin time (placebo group, n=8: 3.2 ± 0.1; dabigatran group, n=9: 6.6 ± 0.5) (Figure 1a).
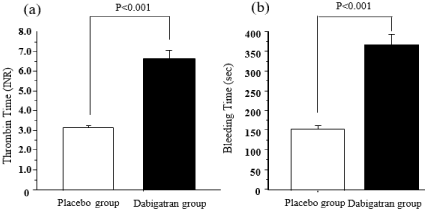
Figure 1: Effects of dabigatran on thrombin time and bleeding time in mice
raised for 22 weeks on a high-fat diet supplemented or not with dabigatran.
(a) Thrombin time: placebo group, n=8; Dabigatran group, n=9. (b) Bleeding
time: placebo group, n=32; Dabigatran group, n=23. Data are presented as
means ± SEM.
Bleeding time
At 14 weeks, bleeding time in the placebo group (n=20) was 161.1 ± 4.9 s and that in the dabigatran group (n=24) was 295.9 ± 16.7 s (data not shown). At 22 weeks, bleeding time in the placebo group (n=32) was 156.4 ± 3.8 s and that in the dabigatran group (n=23) was 367.8 ± 20.0 s (Figure 1b); bleeding time in the dabigatran group was significantly longer than that in the placebo group (P < 0.001).
Development of atherosclerosis
In the placebo and dabigatran groups, we evaluated the extent of atherosclerosis after 22 weeks of feeding (Figure 2). At 22 weeks, the atherosclerotic area in the aorta in the placebo group was 22.8% ± 1.4% (n=9) and that in the dabigatran group was 11.2% ± 0.7% (n=8) (Figure 2b). The atherosclerotic area was significantly (P < 0.001) smaller in the mice given dabigatran than in the mice not given dabigatran.
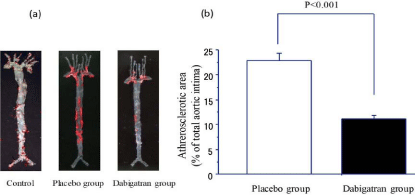
Figure 2: Progression of atherosclerosis in control mice and in mice after
22 weeks of high-fat chow feeding in the placebo and dabigatran groups.
(a) Representative images of atherosclerotic regions identified by Oil Red
O staining of the entire aortic tree. (b) Comparison of atherosclerotic areas
(means ± SEM). Placebo group, n=9; Dabigatran group, n=11.
Histological analysis
Figure 3 shows representative images from a histological analysis of the aortic arch at 22 weeks. Elastica van Gieson staining revealed cleavage of the lamina elastica (arrowing part), medial thickening, and increased collagen fiber in the placebo and dabigatran groups, unlike in the control group (Figure 3a and 3b; control data not shown). Von Kossa staining revealed widespread calcium deposits in the plaque in the placebo group, whereas few calcium deposits were observed in the plaque in the dabigatran group (Figure 3c and 3d).
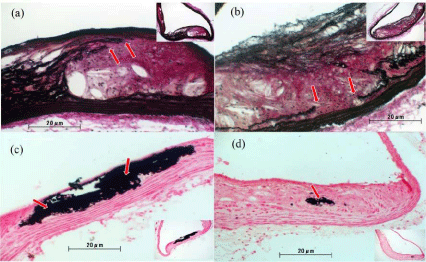
Figure 3: Representative images from histological analysis of the aortic arch
after 22 weeks of experimental chow feeding. (a,c) placebo group (n=6); (b,d)
dabigatran group (n=6). Elastica van Gieson staining (a,b) revealed cleavage
of the lamina elastica in both groups (arrows). Von Kossa staining (c,d)
revealed more widespread calcium deposits in the plaques in the placebo
group than in the dabigatran group (arrows). Magnification: 100× to 200×.
Expression of MMP-9 & VEGF in arteriosclerotic areas
Figure 4 shows representative images from our immunochemical analysis of MMP-9 and VEGF expression in the aortic arch of mice from the placebo and dabigatran groups. Expression levels of MMP-9 (Figure 4a and 4b) and VEGF (Figure 4c and 4d) were significantly lower (MMP-9: P <0.001, n=6; VEGF: P <0.001, n=6) in the dabigatran group than in the placebo group (P=0.0132, n=6) (data not shown).
Expression of t-PA & eNOS in arteriosclerotic areas
Figure 5 shows representative images from immunochemical staining of the aortic arch of mice from the placebo and dabigatran groups for t-PA and eNOS. Expression of t-PA was comparable between the placebo and dabigatran groups (Figure 5a and 5b; P=0.239, n=6). However, expression of eNOS was significantly greater in the dabigatran group than in the placebo group (Figure 5c and 5d; P=0.0132, n=6).
Discussion
Here, we examined the effects of long-term (22 week) direct inhibition of thrombin by dabigatran in Apo-E-/- and LDLR-/--doubleknockout mice to gain insights into the mechanisms underlying the development and progression of atherosclerosis. Our main finding was that oral administration of dabigatran at 7.5mg dabigatran/gchow significantly retarded atherosclerotic lesion development throughout the entire aortic tree (49% decrease) compared with that in placebo mice. In addition, this dose of dabigatran inhibited the long-term progression of atherosclerosis.
Dabigatran has high anticoagulant activity in the plasma of humans and other animal species. Dabigatran inhibits not only the solid-phase thrombin that binds to fibrin but also separated, liquidphase thrombin. The anticoagulant action and the antithrombotic efficacy of dabigatran in vitro and ex vivo are dose dependent [16].
In one study of 18,113 patients with atrial fibrillation, the rate of major bleeding was 3.36% annually in the warfarin-treated group compared with 2.71%-3.11% annually in the dabigatran-treated group (110-150 mg) [17]. In the present study, the treated mice had about the same frequency of hemorrhagic side effects as in humans (data not shown), although it should be noted that we used a high dose here to account for the lower bioavailability of dabigatran in mice than in humans [12]. However, we consider our dose appropriate because the plasma levels of dabigatran in our mice were not higher than the values reported by others [18]. We also observed hemorrhagic side effects in 26-week old mice. Thus, further investigations using senile mice are warranted.
Elastica van Gieson staining revealed rupture of the elastic fibers and the lamina elastica of the plaque and areas of hypertrophy in the tunica media in both the placebo and dabigatran groups (Figure 3a and 3b). In addition, fewer collagen-positive areas were observed in the dabigatran group than in the placebo group, suggesting decreased inflammation in the dabigatran group.
Previous studies have suggested that fibroblast proliferation [19], wound healing and angiogenesis [20]; and induction of inflammatory cytokines [7,8] are inhibited by dabigatran. These findings suggest that the progression of atherosclerosis is accelerated by thrombin.
Von Kossa staining at 22 weeks revealed pathologic findings of Monckeberg medial calcific sclerosis (Figure 3c and 3d) and atheromatous degeneration. In addition, marked arterial calcification was observed, with the calcified lesions being co-localized with plaque in both the placebo and dabigatran groups.
A study in Matrix Gla Protein (MGP)-deficient mice has shown that a decrease of matrix Gla protein induced by administration of vitamin K antagonist leads to acceleration of vascular calcification [21,22]. Warfarin is known to convert vitamin K-dependent coagulation factors to the MGP PIVKA (protein induced by vitamin K absence), thereby indirectly obstructing thrombin generation. However, dabigatran is not a vitamin K antagonist and it is unknown whether dabigatran causes a decrease of MGP expression. Therefore, future studies are needed to examine the action of the thrombin on MGP.
We also examined the expression of VEGF, MMP-9, t-PA, and eNOS by endothelial cells in the aortic arch as indicators of atherosclerosis progression. In atherosclerotic lesions, MMPs, extracellular matrix splitting enzyme, and elastin are involved in plaque rupture. It is thought that atherosclerosis is a restorative response to the damage caused to the intima when the plaque ruptures; that is, it is a rebuilding of the framework of the extracellular matrix in the intima [23]. This suggests that production and degradation of the extracellular matrix must be strictly controlled in the arteriosclerotic nest. Atherosclerotic plaques are a collection of lipids, calcium, and other cellular debris covered by a fibrous cap. Although a large amount of extracellular matrix is present in the fibrous cap, helping to maintain the integrity of the plaque, the resolution of the extracellular matrix induces in the lipid core, and it leads to the fragility of the organization. This fragility causes the plaque to rupture [24]. In both the placebo and dabigatran groups in our study (Figure 4), MMP-9 was found to be localized in endothelial cells in the vicinity of plaque, as well as in the plaque itself; however, significantly less MMP-9 expression was observed in the dabigatran group than in the placebo group. Previous studies have shown that thrombin is involved in activation of the NF-κB pathway, and that thrombin induces upregulation of various adhesion molecules, MMPs (1, 2, 3, 9, 11), cytokines, and growth factors [25,26]. It has also been shown that thrombin increases vasopermeability by inducing the production of MMP-9 [27]. In addition, MMP-9 expression increases if the thrombin is injected into the encephalon basal substance, resulting in cerebral edema [28]. Together, these reports suggest that inhibition of thrombin by dabigatran leads to decreased expression of MMP-9 in atherosclerotic lesions.
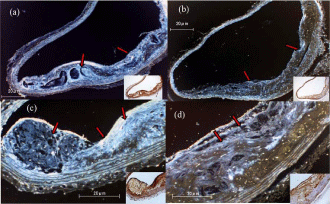
Figure 4: Immunochemistry and pseudo-color treatment of the aortic arch in
the placebo group (a,c) and dabigatran group (b,d) after 22 weeks of feeding.
(a,b) Anti-MMP-9 staining. (c,d) Anti-VEGF staining. Arrows indicate positive
areas. Magnification: 100× to 200×.
VEGF plays a critical role in the development and progression of atherosclerosis by inducing angiogenesis in the induration intima [29]. We therefore examined the expression of VEGF as a pathological index of atherosclerosis progression. In the placebo group, VEGF expression was observed in plaque, especially in foam cells localized in plaque and in smooth muscle cells localized in the lipid-rich core of lesions or immediately below the core. However, significantly less VEGF expression was observed in the dabigatran group. The progression of atherosclerosis is known to involve migration and transformation of vascular smooth muscle cells; this is induced by chemokines and growth factors, including VEGF, secreted by macrophages and foam cells [30]. Thrombin is thought to promote the expression of VEGF by a protease activated receptor-1-mediated mechanism and the induction of chemokine secretion. Thrombin is also involved in the hypoxia-inducible factor-1a signal transmission pathway, which is involved in both VEGF gene expression and angiogenesis [31]. Together, these findings suggest that dabigatranmediated inhibition of these activities of thrombin leads to decreased expression of VEGF.
t-PA expression was observed in regions of endothelium that were associated with plaque in both the placebo and dabigatran groups, and the expression levels did not differ between the groups (Figure 5a and 5b). Therefore, thrombin inhibition had no effect on t-PA levels (data not shown), suggesting that thrombin has no directly effect on t-PA expression.
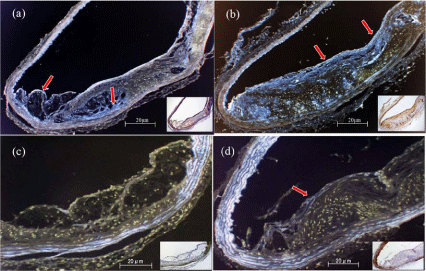
Figure 5: Immunochemistry and pseudo-color treatment of the aortic arch in
the placebo group (a,c) and dabigatran group (b,d) after 22 weeks of feeding.
(a,b) Anti-t-PA staining. (c,d) Anti-eNOS staining. Red arrows indicate
positive areas. Magnification: 100× to 200×.
eNOS expression was observed in vascular endothelial cells in the dabigatran group but not in the placebo group (Figure 5c and 5d). This expression was particularly low in plaque-associated regions of endothelium. The lack of eNOS expression in the placebo group suggests that the production of NO, which is necessary for homeostasis of the microcirculation [32], was diminished in atherosclerotic regions. Thrombin induces the expression of iNOS and increases inflammation [33]. Generation of superoxides, leading to the production of peroxynitrite and hydrogen peroxide, would likely have a greater effect on the progression of atherosclerosis than would the generation of NO. Dabigatran-induced thrombin inhibition might prevent the effects of these compounds on the progression of atherosclerosis. The influence of thrombin inhibition on the expression of iNOS should be assessed in future experiments.
In summary, long-term (22 weeks) oral administration of the thrombin inhibitor dabigatran in ApoE-/- and LDLR-/- doubleknockout mice led to significant inhibition of atherosclerosis progression and to significant decreases in expression of the progression-related mediators MMP-9 and VEGF. These results suggest that thrombin plays an important role in the progression of atherosclerosis. We also found that dabigatran increased the expression of eNOS, suggesting an additional role of thrombin in preventing eNOS expression. Further studies examining the multiple biological functions of thrombin and the roles of thrombin in the progression of atherosclerosis, as well as examining how thrombin inhibition by dabigatran influences the progress of atherosclerosis, are warranted.
Acknowledgements
Placebo chow and dabigatran etexilate–supplemented chow were kindly donated by Boehringer Ingelheim (Ingelheim am Rhein, Germany).
References
- Lippi G, Franchini M and Targher G. Arterial thrombus formation in cardiovascular disease. Nat Rev Cardiol. 2011; 8: 502-512.
- Jennings LK. Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009; 102: 248-257.
- Bogatcheva NV, Garcia JG and Verin AD. Molecular mechanisms of thrombininduced endothelial cell permeability. Biochemistry (Mosc). 2002; 67: 75-84.
- Hatake K, Kakishita E, Wakabayashi I, Sakiyama N and Hishida S. Effect of aging on endothelium-dependent vascular relaxation of isolated human basilar artery to thrombin and bradykinin. Stroke. 1990; 21: 1039-1043.
- Wang HJ, Chen SF and Lo WY. iTRAQ quantitative proteomics-based identification of cell adhesion as a dominant phenotypic modulation in thrombin-stimulated human aortic endothelial cells. Thromb Res. 2015; 135: 944-950.
- Grover SP and Mackman N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler Thromb Vasc Biol. 2018; 38: 709-725.
- Naldini A, Carney DH, Bocci V, Klimpel KD, Asuncion M, Soares LE, et al. Thrombin enhances T cell proliferative responses and cytokine production. Cell Immunol. 1993; 147: 367-377.
- Sower LE, Froelich CJ, Carney DH, Fenton JW, 2nd and Klimpel GR. Thrombin induces IL-6 production in fibroblasts and epithelial cells. Evidence for the involvement of the Seven- Transmembrane Domain (STD) receptor for alpha-thrombin. J Immunol. 1995; 155: 895-901.
- Larsen TB, Skjoth F, Nielsen PB, Kjaeldgaard JN and Lip GY. Comparative effectiveness and safety of non-vitamin K antagonist oral anticoagulants and warfarin in patients with atrial fibrillation: propensity weighted nationwide cohort study. BMJ. 2016; 353: i3189.
- Hankey GJ and Eikelboom JW. Dabigatran etexilate: a new oral thrombin inhibitor. Circulation. 2011; 123: 1436-1450.
- Kakar P, Watson T and Lip GY. Drug evaluation: rivaroxaban, an oral, direct inhibitor of activated factor X. Curr Opin Investig Drugs. 2007; 8: 256-265.
- Stangier J and Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost. 2009; 15: 9S-16S.
- Stangier J, Rathgen K, Stahle H, Gansser D and Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol. 2007; 64: 292-303.
- Yamashita T, Oda E, Sano T, Ijiru Y, Giddings JC and Yamamoto J. Varying the ratio of dietary n-6/n-3 polyunsaturated fatty acid alters the tendency to thrombosis and progress of atherosclerosis in apoE-/- LDLR-/- double knockout mouse. Thromb Res. 2005; 116: 393-401.
- Suzuki A, Togashi K, Nokubi M, Koinuma K, Miyakura Y, Horie H, et al. Evaluation of venous invasion by Elastica van Gieson stain and tumor budding predicts local and distant metastases in patients with T1 stage colorectal cancer. Am J Surg Pathol. 2009; 33: 1601-1607.
- Wienen W, Stassen JM, Priepke H, Ries UJ and Hauel N. Effects of the direct thrombin inhibitor dabigatran and its orally active prodrug, dabigatran etexilate, on thrombus formation and bleeding time in rats. Thromb Haemost. 2007; 98: 333-338.
- Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, et al. Committee R-LS and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009; 361: 1139-1151.
- Fusaro M, Dalle Carbonare L, Dusso A, Arcidiacono MV, Valenti MT, Aghi A, et al. Differential Effects of Dabigatran and Warfarin on Bone Volume and Structure in Rats with Normal Renal Function. PLoS One. 2015; 10: e0133847.
- Perdue JF, Lubenskyi W, Kivity E, Sonder SA and Fenton JW 2nd. Protease mitogenic response of chick embryo fibroblasts and receptor binding/ processing of human alpha-thrombin. J Biol Chem. 1981; 256: 2767-2776.
- Carney DH, Mann R, Redin WR, Pernia SD, Berry D, Heggers JP, et al. Enhancement of incisional wound healing and neovascularization in normal rats by thrombin and synthetic thrombin receptor-activating peptides. J Clin Invest. 1992; 89: 1469-1477.
- Poterucha TJ and Goldhaber SZ. Warfarin and Vascular Calcification. Am J Med. 2016; 129: 635.
- Caluwe R, Pyfferoen L, De Boeck K and De Vriese AS. The effects of vitamin K supplementation and vitamin K antagonists on progression of vascular calcification: ongoing randomized controlled trials. Clin Kidney J. 2016; 9: 273-279.
- Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999; 340: 115-126.
- Falk E, Shah PK and Fuster V. Coronary plaque disruption. Circulation. 1995; 92: 657-671.
- Machida T, Takata F, Matsumoto J, Takenoshita H, Kimura I, Yamauchi A, et al. Brain pericytes are the most thrombin-sensitive matrix metalloproteinase-9- releasing cell type constituting the blood-brain barrier in vitro. Neurosci Lett. 2015; 599: 109-114.
- Borissoff JI, Spronk HM, Heeneman S and ten Cate H. Is thrombin a key player in the ‘coagulation-atherogenesis’ maze? Cardiovasc Res. 2009; 82: 392-403.
- Kawakita K, Kawai N, Kuroda Y, Yasashita S and Nagao S. Expression of matrix metalloproteinase-9 in thrombin-induced brain edema formation in rats. J Stroke Cerebrovasc Dis. 2006; 15: 88-95.
- Liu DZ, Ander BP, Xu H, Shen Y, Kaur P, Deng W, et al. Blood-brain barrier breakdown and repair by Src after thrombin-induced injury. Ann Neurol. 2010; 67: 526-533.
- Zhu Y, Carmeliet P and Fay WP. Plasminogen activator inhibitor-1 is a major determinant of arterial thrombolysis resistance. Circulation. 1999; 99: 3050- 3055.
- Torr-Brown SR and Sobel BE. Attenuation of thrombolysis by release of plasminogen activator inhibitor type-1 from platelets. Thromb Res. 1993; 72: 413-421.
- Vercauteren E, Peeters M, Hoylaerts MF, Lijnen HR, Meijers JC, Declerck PJ, et al. The hyperfibrinolytic state of mice with combined thrombin-activatable fibrinolysis inhibitor (TAFI) and plasminogen activator inhibitor-1 gene deficiency is critically dependent on TAFI deficiency. J Thromb Haemost. 2012; 10: 2555-2562.
- Moncada S and Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol. 2006; 213-254.
- Kang KW, Choi SY, Cho MK, Lee CH and Kim SG. Thrombin induces nitricoxide synthase via Galpha12/13-coupled protein kinase C-dependent I-kappa B-alpha phosphorylation and JNK-mediated I-kappa B-alpha degradation. J Biol Chem. 2003; 278: 17368-17378.