
Review Article
Thromb Haemost Res. 2022; 6(3): 1081.
Laboratory Diagnosis and Classification of Von Willebrand Disease: A Review
Vangenechten I1,2,3* and Gadisseur A1,2,3,4
1Haemostasis Unit, Department of Haematology, Antwerp University Hospital, Edegem, Belgium
2Haemostasis Research Unit, Antwerp University, Antwerp, Belgium
3Department of Haematology, Antwerp University Hospital, Edegem, Belgium
4CSL Behring Chair in von Willebrand Disease, Antwerp University, Antwerp, Belgium
*Corresponding author: Inge Vangenechten, Haemostasis Unit, Department of Haematology, Antwerp University Hospital1, Drie Eikenstraat 655, B - 2650 Edegem, Belgium
Received: August 22, 2022; Accepted: September 23, 2022; Published: September 30, 2022
Abstract
Von Willebrand Disease (VWD), as the most common inherited bleeding disorder, is a widely misdiagnosed disease due to several diagnostic pitfalls. At the heart of this challenge lies the complexity and heterogeneity of VWF, significant (pre-) analytic issues, limited access to a comprehensive repertoire of laboratory assays, inter-individual variations, lack of expertise and complex interpretation of results. Next to a personal and family bleeding history, an array of clinical laboratory tests is required because no single test reflects both quantity and quality of VWF. The assays measure different VWF properties and may be affected by (pre-) analytic variables possibly leading to inaccurate interpretation. Therefore, a laboratory investigation and diagnosis according to a standard algorithm, and repetitive testing, are essential for accurate diagnosis which is illustrated in detail in this review.
Keywords: Classification and diagnosis; Laboratory assays; Von willebrand disease; Von willebrand factor activity; Von willebrand factor antigen
Introduction
Although it is the most common autosomal inherited bleeding disorder, Von Willebrand Disease (VWD) is an under diagnosed and relatively unknown disease with his accurate diagnosis to be very challenging. A panel of laboratory assays measuring different VWF functions is used in diagnosis and classification of VWD. Introduction of new improved techniques can contribute to better diagnosis, classification and treatment. This review describes the laboratory assays for VWD diagnosis and classification, including their mechanisms and pitfalls.
Von Willebrand Disease
VWD is characterized by mucocutaneous bleeding, like bruising, epistaxis and menorrhagia [1], prolonged bleeding after trauma and surgery, and occurs with equal frequency among man and women. The estimated prevalence is 0.1 – 1.0%, although actual figures may be higher since a large number of people with a VWF mutation are asymptomatic or undiagnosed. Different from Hemophilia, VWD is caused by a deficiency or dysfunction of Von Willebrand Factor (VWF), giving rise to deficits both in primary and plasmatic haemostasis.
VWF Structure
The VWF gene is located at chromosome 12p13.3 compromising 52 exons [2], and mutations located in different domains of the VWF protein [3] give rise to VWD with domain-specific function abnormalities. The intracellular uncleaved VWF has a domain structure constructed as follows: SP-D1-D2-D’-D3-A1-A2-A3-D4- C1-C2-C3-C4-C5-C6-CK [3] (Figure 1). D1-D2 represents the VWF propeptide and are cleaved of during proteolytic processing [4]. The remaining domains form the mature VWF with their own specific function; binding of factor VIII (D’-D3), platelet Glycoprotein (GP) Ib receptors (A1) or Collagen (A1 and A3), and ADAMTS-13 cleavage site (A2 at position Tyr1605-Met1606).
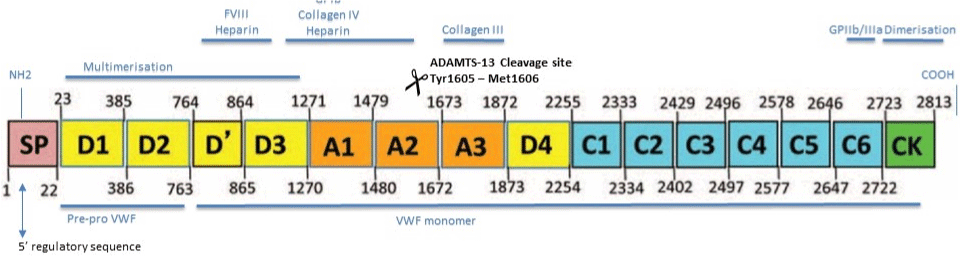
Figure 1: VWF gene structure.
Abbreviation: FVIII: Factor VIII; GPIb: Glycoprotein Ib; GPIIb/IIIa: Glycoprotein IIb/IIIa
VWF Functions
At sites of vascular injury, VWF binds to sub endothelial collagen and platelet GPIb, allowing platelet adhesion to these sites. This VWF-platelet interaction is needed in areas of high shear stress where direct platelet-collagen binding is ineffective. It also promotes platelet-platelet interactions (aggregation) by binding platelet membrane GPIIb/IIIa.
Additionally, VWF protects coagulation factor VIII from rapid proteolytic degradation and exposes it to the site of vascular damage, thus indirectly also contributing to the plasmatic coagulation process [5].
This multifunctional nature of VWF contributes to the heterogeneity in clinical symptoms and bleeding risk, but also poses a diagnostic challenge in VWD.
VWD Classification
Because there is no clear correlation between bleeding severity and a particular laboratory test, VWD classification is not straightforward. The current International Society on Thrombosis and Haemostasis (ISTH)/Scientific and Standardization Committee (SSC) classification [6] (Table 1) is based on a limited panel of laboratory parameters (coagulation factor VIII (FVIII:C), VWF antigen (VWF: Ag) and VWF Ristocetin Cofactor activity (VWF: RCo), Ristocetin Induced Platelet Aggregation (RIPA) and VWF Multimeric analysis (VWF: MM))allowing classification of VWD into 3 main categories: type 1 (partial quantitative deficiency), type 2 (qualitative defect) and type 3 (complete deficiency).
![]()
FVIII:C
VWF:Ag
VWF:RCo
VWF:RCo/VWF:Ag
VWF:CB
VWF:CB/VWF:Ag
VWF mutlimers pattern
Specializedassays
VWF gene mutation
Type 1
N/↓
↓
↓
= 0.60
↓
= 0.60
N*
Whole gene
Type 1C
N/↓
↓
↓
= 0.60
↓
= 0.60
N
VWFpp/VWF:Ag ↑
D3
Type 3
↓↓↓
↓↓↓
↓↓↓
↓↓↓
absent
Whole gene
Type 2A
N/↓
↓
↓↓
= 0.60
↓↓
= 0.60
Loss of
A2
HMWM/IMWM
D2
D3
CK
Type 2B
N/↓
↓
↓↓
= 0.60
↓↓
= 0.60
Loss of
LD-RIPA ↑
A1
HMWM
Type 2M
N/↓
↓
↓↓
= 0.60
N
= 0.60
N*
A1
Type 2N
↓↓
N/↓
N/↓
= 0.60
N/↓
= 0.60
N
VWF:FVIIIB ↓
D’-D3
↓, mild decrease; ↓↓, moderate decrease; ↓↓↓, severe decrease; ↑, increase; HMWM, high molecular weight multimers; IMWM, intermediate molecular weight multimers;N, normal. *subtle changes are allowed [42,43].
Table 1: Laboratory phenotype of different VWD types.
Type 1, affecting>70% of all VWD patients, is characterized by a partial reduction of a functionally normal VWF, with a normal VWF activity/VWF: Ag ratio and the presence of all VWF multimers. A type 1 subtype, type 1 Vicenza (recently renamed as type 1 clearance (1C)) is characterized by a significant increased VWF clearance, resulting in low VWF levels and a good but short lived response to Desmopressin.
There is some controversy in type 1 VWD versus “Low VWF” [7]. It has been suggested to reserve type 1 diagnosis for patients with VWF levels <30IU/dL [8,9] and using the term “low VWF” when VWF level is 30-50IU/dL [10,11]. In this last group there is a low association with mutations and using “Low VWF” avoids misdiagnosis of these patients as having an inherited bleeding disorder. This has recently been reassessed and currently diagnosis of VWD can be made with VWF 30-50IU/dL with a history of abnormal bleeding [9].
Type 2, 20% of all VWD cases, is divided into four subtypes (2A, 2B, 2M and 2N) according to VWF functional defects (Table 1). The Antwerp Haemostasis Research Unit (University Hospital Antwerp) and the CSL Behring Chair in von Willebrand disease at Antwerp Universityuse an extended ISTH/SSC classification [6] with additional subdivision of type 2A into 2A/IIA, IIC,IID and IIE [12,13], based on VWF multimeric patterns and unique domain location of the VWF mutation (Table2).
![]()
FVIII:C
VWF:Ag
VWF:RCo
VWF:RCo/VWF:Ag
VWF:CB
VWF:CB/VWF:Ag
VWF mutlimerspattern
Specializedassays
VWF gene mutation
Quantitative defect of VWF
Type 1
N/↓
↓
↓
= 0.60
↓
= 0.60
N*
Whole gene
Type 1C
N/↓
↓
↓
= 0.60
↓
= 0.60
N
VWFpp/VWF:Ag ↑
D3
Type 3
↓↓↓
↓↓↓
↓↓↓
↓↓↓
absent
Whole gene
Qualitative defect of VWF
Type 2A/IIA
N/↓
↓
↓↓
= 0.60
↓↓
= 0.60
↓↓↓ HMWM+IMWM
A2
Enhanced proteolytic bands
Type 2A/IIC
N/↓
↓
↓↓
= 0.60
↓↓
= 0.60
↓↓ of HMWM
D2
Increased protomer
Decrease of satellite bands
Type 2A/IID
N/↓
↓
↓↓
= 0.60
↓↓
= 0.60
↓ of HMWM
CK
Extra central bands
Type 2A/IIE
N/↓
↓
↓↓
= 0.60
↓↓
= 0.60
↓↓ of HMWM
D3
Loss of outer proteolytic bands
Type 2B
N/↓
↓
↓↓
= 0.60
↓↓
= 0.60
↓↓ of
LD-RIPA ↑
A1
HMWM
Type 2M
N/↓
↓
↓↓
= 0.60
N
= 0.60
N*
A1
Type 2N
↓↓
N/↓
N/↓
= 0.60
N/↓
= 0.60
N
VWF:FVIIIB ↓
D’-D3
↓, mild decrease; ↓↓, moderate decrease; ↓↓↓, severe decrease; ↑, increase; HMWM, high molecular weight multimers; IMWM, intermediate molecular weight multimers;N, normal. *subtle changes are allowed [42,43].
Table 2: Laboratory phenotype of different VWD subtypes.
Type 3, a rare form accounting for<5% of all VWD cases, is characterized by a near complete absence of VWF: Ag and VWF: RCo (<1IU/dL) and VWF propeptide levels (<5IU/dL) [14], whereas severe type 1 has low VWF: Ag levels but nearly normal VWF propeptide levels (>5IU/dL) [15]. The almost absent VWF gives rise to a non-stabilized FVIII resulting in low FVIII: C levels (<5IU/dL). This type is caused by a homozygous or compound heterozygous inheritance [16].
Laboratory Diagnosis of VWD
Clinically VWD can be suspected when there is history of mucocutaneous bleeding symptoms or bleeding during surgery, which triggers laboratory analysis.
The current diagnostic approach is based upon a few routinely available screening tests; VWF: Ag, VWF platelet Glycoprotein (GP) Ib binding function and FVIII:C because of its relationship with VWF. These tests allows for VWD classification into his three main types (type 1, 2 and 3).
If available, other functional tests can be added to further investigate type 2; VWF binding to collagen (VWF: CB) or FVIII (VWF: FVIIIB), RIPA and VWF:MM. Rapidly gaining interest is the VWF propeptide measurement reflecting VWF protein production, secretion and elimination.
Most laboratories make use of an algorithm of all available assays for the investigation of suspected VWD and classification [17] (Figure 2).
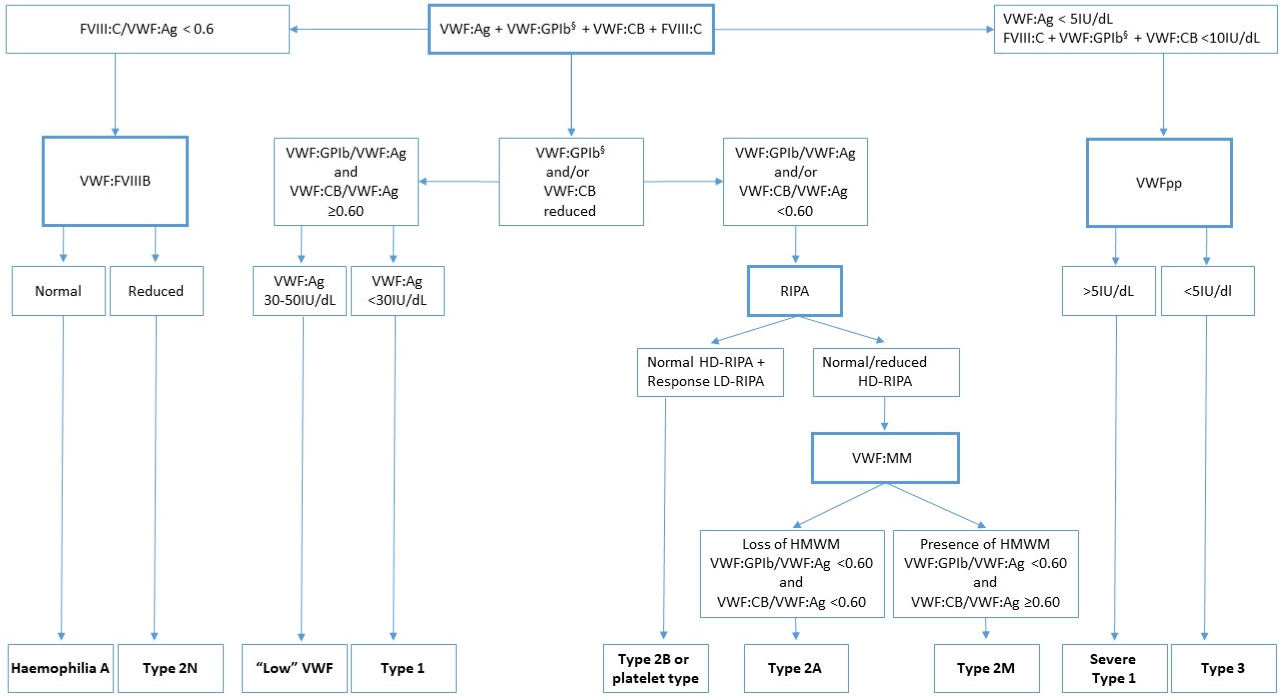
Figure 2: A diagnostic algorithm that describes the VWD diagnostic and classification process using laboratory testing.
Abbreviations: Ag: Antigen; CB: Collagen Binding; FVIII: Factor VIII; GPIb: Glycoprotein Ib; HD: High Dose; HMWM: High Molecular Weight Multimers; LD: Low
Dose; RIPA: Ristocetin Induced Platelet Aggregation; VWF: Von Willebrand Factor; VWD: Von Willebrand Disease
VWF Antigen
The VWF Antigen (VWF: Ag) quantification, determining VWF protein plasma levels but not its functional status, is most often the first diagnostic step. It is typically done using an ELISA (enzymelinked immunosorbent assay), ora more rapid automated LIA-test (latex particle immune turbidimetric assays). The newer CLIA (chemiluminescent immunoassay; HemosIL® AcuStar® VWF:Ag, Werfen, Barcelona, Spain) based assay shows good agreement with the currently used assays. It offers the lowest assay variation and a Lower Limit Of Quantification (LLOQ) of 0.2IU/dL compared with the other VWF antigen assays; LIA: 0-5IU/dL and ELISA: 5-10IU/ dL [18].
An automated LIA VWF: Ag assay is preferred due to its excellent overall performance, stability, single determinations and precision. Unfortunately, the CLIA assay is restricted to the AcuStar®.
Factor FVIII Activity
The FVIII activity (FVIII: C) is a clot based one-stage assay measuring the ability to correct the Activated Partial Thromboplastin Time (APTT) of FVIII deficient plasma, compared with a normal plasma.
VWF is a carrier of FVIII in the circulation forming a VWF/ FVIII complex in a 1:1 ratio in healthy individuals, blood group independent. This ratio (FVIII: C/VWF: Ag) can distinguish normal subjects from VWD type 2N because of a significant loss of FVIII:C caused by a FVIII binding defect of VWF.
The FVIII: C assay is a rapid, simple to perform, automated assay which is widely used for clinical monitoring.
The assay is influenced by large variation of assay kits/reagents, assay conditions and which analysers are used which may result in high inter-laboratory variability.
VWF – Platelet Dependent Binding Activity
VWF platelet dependent binding activity assays (VWF: GPIb) measure the capacity of VWF to bind GPIb platelet receptors and allow to distinguish type 1 and 2 VWD, and differentiation of type 2 into type 2A/B and 2M VWD.
Historically, the VWF Ristocetin Cofactor assay (VWF: RCo) using manual light transmission aggregometry (LTA) was the “gold” standard method. It measures VWF-dependent agglutination in the presence of ristocetin, an antibiotic mediating the VWF binding to platelet GPIb receptor [19]. Because the VWF: RCo was impacted by many issues leading to VWD misdiagnosis (time-consuming, difficult execution and interpretation, LLOQ of 10-20IU/dL insufficiently low to distinguish dominant type 1 versus type 2M versus 2A/IIE, high intra- and inter-laboratory coefficients of variation (CV’s) of 25- 30IU/dL, and reproducibility issues), the assay needed to be repeated several times to confirm (low) levels.
Currently, automated LIA-assays, developed to improve precision and LLOQ of VWF: GPIb detection, with or without ristocetin, are widely used. They are all based on optical density changes provoked by micro particle agglutination directly proportional to the VWF:GPIb activity amount (Table 3) [20].
![]()
Assay
ISTH nomenclature
principles
VWF:RCo by aggregometry “gold” standard
VWF: RCo
Plasma VWF aggregates/agglutinates with washed/lyophilized platelets in the presence of ristocetin
HemosIL VWF activity
VWF: Ab
VWF agglutinates with particles coated with a specific anti-VWF mouse mAb that binds the binding site of platelet GPIb receptor (VWF A1 domain), without the requirement of ristocetin
HemosIL VWF:RCo
VWF: GPIbR
VWF agglutinates with latex particles coated with a monoclonal antibody (mAb) to measure VWF binding to recombinant GPIb fragments (rfGPIba) in presence of ristocetin
Innovance VWFAc
VWF: GPIbM
VWF agglutinates spontaneously with latex particles containing two gain-of-function mutations (G233V, M239V)
HemsoIL Acustar VWF: RCo
VWF: GPIbR
VWF agglutinates with magnetic beads coated with recombinant GPIb in the presence of ristocetin and detected by isoluminol labeled anti-VWF antibodies
Abbreviations: GPIb: Glycoprotein Ib; RCo: Ristocetin Cofactor; VWF: Von Willebrand Factor.
Table 3: VWF GPIb binding activity assays.
The HemosIL®VWF: RCo assay [21] (VWF: GPIbR, Werfen) uses a monoclonal antibody (mAb) to measure VWF binding to recombinant GPIb fragments (rfGPIba) coated onto latex particles in presence of ristocetin. In contrast, the HemosIL® VWF activity assay [22] (VWF: Ab, Werfen) uses particles coated with a specificant i-VWF mousem Ab that binds the binding site of platelet GPIb receptor, without the requirement of ristocetin. The Innovance® VWF: Ac assay [23] (VWF: GPIbM, Siemens Healthcare Diagnostics, Marburg, Germany ) utilizes a rfGPIba, containing two gain-offunction mutations (G233V, M239V),which bind plasma VWF through its GPIb receptor spontaneously.
All three methods have shown to have lower LLOQ (respectively 4, 3 and 2IU/dL), improved accuracy to distinguish type 1 from 2M, even as low as 3-30IU/dL, and better inter- and intra-assay variation (<10%) compared with the traditional VWF: RCo [22,24]. Recently a CLIA based assay (HemosIL® AcuStar® VWF: RCo, Werfen) [25] has become available offering an even lower LLOQ of 0.2IU/dL and an imprecision of <8%, even in the lower level range [26].
By using the ristocetin dependent assays, significant lower VWF: GPIb levels were seen in carriers of specific VWF amino acid polymorphisms (i.e. D/H1472) compared to wild type VWF [27]. Although this polymorphism has no clinical significance, it can lead to VWD misdiagnosis. In contrast, the gain of function GPIb assay (VWF: GPIbM) did not show any difference between D/H1472 carriers and non-carriers, probably due to a minor affinity of VWF A1 domain to bind ristocetin [27].
Based on our comparative analysis on different automated VWF: GPIb activity assays in a well-defined VWD cohort [28], the VWF: GPIbM seemed to be the best choice as it provides the best balance between sensitivity and specificity for type 2, the lowest LLOQ compared with VWF: Ab and VWF: GPIbR, and is not affected by polymorphisms giving false low VWF:GPIb levels. Despite the even lower LLOQ of the CLIA based assay, the VWF: GPIbM is preferred as it does not require a specific coagulation analyser, where the CLIA assay is restricted to the AcuStar® (Werfen). It has to be noted that even though these assays measure VWF: GPIb, they are not a “true” VWF activity assay because they determine an antigen-antibody reaction rather than a VWF-platelet binding as is the case in VWF: RCo by aggregometry.
Platelet Function Assay
The Platelet Function Analyzer (PFA-100, Siemens) using Collagen/ADP and Collagen/Epinephrine cartridges as screening tool for abnormalities in primary haemostasis [29], is easy to perform and uses whole blood. It measures the closure time (CT, occlusion of blood flow) in a shear flow environment after blood is exposed to the agonist (COL/ADP and/or COL/EPI) coated membrane within the cartridge. It has a high negative predictive value, i.e. with some exceptions (Storage Pool Disease, Primary Secretion Defects, mild Type 1 VWD) normal PFA results correlate with an intact primary haemostasis. Prolonged CT are less specific arising from VWD, platelet dysfunction, low platelet count or low hematocrit. Generally in more significant VWD both COL/EPI and COL/ADP CT will be prolonged, where this may be only the case for COL/EPI in milder forms. The PFA-100 is very sensitive to the presence or absence of VWF; >99% sensitive to VWD type 2A, 2B, 2M and type 3; and around 80% sensitive to VWD type 1 (increasing sensitivity ~decreasing VWF) [30].
In conclusion, in normal practice, VWD and subtypes can be determined by a broad VWD test panel (VWF: Ag, VWF: GPIb, FVIII:C and PFA) but enormous variations between hospitals and a large degree of misdiagnosis/-classification can be due to the performance of insufficient test panels or inappropriate tests [31]. The performance of one single assay is not sufficient to diagnose/ classify VWD. (E.g. PFA-100 results reflect abnormalities in primary haemostasis not restricted to VWD. By using only VWF: Ag, type 2 VWD can be missed, as normal VWF: Ag levels are seen in type 2. Therefore, for a satisfactory analysis, laboratories should employ a comprehensive panel [32] that comprises FVIII, VWF: Ag, VWF: GPIb and PFA which can help clinicians to detect the presence of quantitative or qualitative VWF defects and/or determine the need for further investigation.
Specialized Laboratory Assays
Once VWD is diagnosed, additional assays, mostly only provided in specialized (reference) laboratories, can be used. This will provide additional information for classification of VWD into different subtypes.
VWF Collagen Binding
The VWF collagen binding assay (VWF: CB) was developed out of concerns over sensitivity, reproducibility and inter-laboratory variability of VWF:R Cobinding assay, and was initially proposed as replacement for the latter because of less inter-assay and interlaboratory variability. However, it measures another VWF function than platelet binding [33], namely the binding of VWF to collagen mostly by the A3 domain (major binding site of collagen type III/I), while GPIb binding happens in the A1 domain. The ability of VWF to bind collagen is associated with High Molecular Weight Multimers (HMWM) [34], which are also important for platelet adhesion. Specific mutations in the A3 domain [35] affect the ability of VWF to bind type I/III collagen. Additionally, patients with VWF: CB defects may have normal VWF: GPIb activities. This would only be picked up by using the VWF: CB assay.
The source and types of collagen are important variables [36]. A collagen mixture of type I/III collagen from equine or bovine tendon is generally better to detect HMWM in contrast with purified animal derived type I collagen or human derived type III collagen. Collagen type I seems to bind poorly with VWF, while type III collagen will bind too well. The VWF: CB assay is an ELISA which mainly uses type I/III collagen [37]. A minor collagen binding site (collagen type IV and VI) is present in the A1 domain of the VWF gene which binds to different sites on collagen [35,38], but these assays are not widely available or used.
There is an increasingly interest for the automated CLIA assay (HemosIL® AcuStar VWF: CB, Werfen) which uses type III collagentriple- helical peptide coated magnetic particles. This new technique has major advantages; it is rapid, simple in use, ready-to-use reagent cartridges and can be an addition for non specialized laboratories. It shows high sensitivity to low VWF levels and type 2 versus type 1 VWD although with a lower discrimination between type 2A and 2M [39]. Although theoretically this assay has important advantages especially in rapidity, the assay can only be used on the AcuStar®.
VWF Activity to Antigen Ratios
VWF: GPIb/VWF: Ag ratio was introduced to differentiate VWD type 1 from 2, with a ratio ~1.0 as denoting type 1 and a reduced ratio for type 2 (except 2N). The VWF: CB/VWF: Ag ratio has been considered to reflect a multimer distribution outside the reference range interval [40].
Using both ratios, it is possible to distinguish type 1 from type 2A/B and type 2M, without distinction between 2A and 2B, which is usually done by RIPA. In type 1 and 2A/B, both ratios are respectively normal and reduced. In general, type 2M patients are characterized with a reduced VWF: GPIb/VWF: Ag ratio with normal VWF: CB levels. However, a very rare type 2M with reduced VWF: CB and normal VWF: GPIb levels has been described [41], called type 2M-collagen binding defect VWD.
The cut-off ratio to define type 2 has been variously set at <0.70 [9,42], <0.60 [43] and even lower at <0.50 in some studies, but based on our own experience, we recommend to use the 0.60 cut-off, as it is advocated by several publications and the British guidelines [8,14,43]. It has to be noted that with important laboratory CV’s in all three tests (VWF: Ag; VWF: GPIb and VWF: CB) these ratios may not be reliable and differ from blood sampling to blood sampling. In case of VWF: Ag and/or VWF GPIb and/or VWF: CB levels <15IU/dL, the ratios must not be used because they become too unreliable.
Ristocetin Induced Platelet Aggregation
Ristocetin Induced Platelet Aggregation (RIPA) is an ex-vivo platelet function assay which measures platelet aggregation with the help of VWF and exogenous ristocetin in low and high dose as agonist (0.6 and 1.2 mg/mL respectively) [44]. Ristocetin causes exvivo formation of the A1 loop whereby VWF binds to platelet GPIb.
In general, RIPA will identify severe or moderate VWD by the lack of aggregation at high dose RIPA. However, “low VWF” individuals (levels 30-50IU/dL) may have normal results. Compared to the currently used VWF: GPIb assays, RIPA has an added benefit in diagnosis of type 2B/pseudo VWD because it uses patient’s endogenous platelets, whereas VWF: GPIb assays uses exogenous fixed platelets to evaluate the VWF function and not the platelet function.Therefore, the most important role of RIPA is the accurate type 2B identification because this impacts prognosis and treatment. In type 2B,with higher platelet binding affinity, Desmopressin treatment will increase the abnormal VWF, resulting in further binding of platelets and development of thrombocytopenia and potentially greater bleeding making Desmopressin contraindicated in case of suspected type 2B.
Evaluation of RIPA by Light Transmission Aggregometry (LTA), performed on Platelet Rich Plasma (PRP), is considered the “gold” standard [45]. It determines platelet aggregation by measuring light transmission increases in response to addition of the platelet agonists to the platelet suspension. Making PRP and assay execution is very time consuming, labor-intensive, is affected by different pre-analytic conditions (i.e. low platelet count, lipemic plasma) and requires some expertise to interpret the results. The test can only be done on fresh samples. This all limits the use of LTA to specialized laboratories.
As a consequence, Whole Blood (WB) impedance aggregometry, which is time-sparing and needs less blood, has been introduced in more routine laboratories (i.e. Multiplate® system Roche Diagnostics, Mannheim, Germany). There is no need for centrifugation. The test is based on impedance changes proportional to the number of platelets sticking to two electrodes through which an alternating current is passed; results expressed in arbitrary units corresponding to the area under the aggregation curve observed over time. The WB-RIPA on Multiplate is recommended as RIPA assay for reliable differentiation between VWD (type 2B) and non-VWD, with as few operational steps as possible, more time-saving, and without the need of an expert interpretation and has a potential role as a rapid VWD screening test [46].
VWF Multimer Assay
VWF circulates in plasma in different multimeric forms of different molecular weight with the HMWM having the greatest haemostatic capacity. Undergoing ADAMTS-13 mediated proteolysis, under high sheer stress conditions the initial VWF multimer distribution is remodeled by converting large multimers into smaller parts with production of cleavage products, all leading to typical steady-state electrophoresis patterns which vary from mutation to mutation.
The VWF multimeranalysis (VWF: MM) is a specialized technique which evaluates the size distribution of multimers and aids in VWD subtype differentiation. It is the ultimate arbiter for the distinction between type 1, type 2M (both traditionally defined as having normal multimers, although subtle changes have been allowed [42,43]) and type 2 VWD with abnormal multimers (type 2A variants, 2B).
The “gold” standard multimer technique [47], not routinely available, allows a sensitive and precise analysis of multimeric patterns using different resolution gels (0.6-1.2%). The visual number of bands is counted by naked eye. Theoretically, quantification using densitometric curves is possible but not reliable because of standardisation issues due to differences between gels, technical skills, etc. which may lead to incorrect VWD subtyping. Expert knowledge is always required. Only a limited number of centers have been able to duplicate this technique successfully.
These mi-automated assays, HYDRAGEL VW multimers (Sebia, Lisses, France) is more standardised with a ready to use kit and is performed on a single instrument which reduces workload and gives results within 1-day. It is reliable, demonstrating comparable results with the “gold” standard technique with an acceptable discrimination between type 1 and 2 VWD. It has been shown that the HYDRAGEL offers the possibility of quantification of densitometry by calculating the Area Under the Curve (AUC), which standardizes a “visual” technique with pathological reference ranges that can simplify VWD classification, making it less observer dependent [48]. The HYDRAGEL VW multimers assay can be used in routine laboratories to detect HMWM losses but is restricted to the HYDRASYS 2 SCAN analyser. So, unless it is already accessible in the lab as a multiparameter gel electrophoresis system, it will need to be especially acquired.
This being said, VWF multimers do not protect against errors in VWD diagnosis/classification. In a series of ‘type 1 VWD’ studies [49], based on ‘diagnosis’ of type 1 VWD using VWF: Ag, VWF: RCo and VWF: MM around 15-20% of type 1 cases were later reidentified as type 2 (predominantly 2M) VWD. Therefore it is of great importance that VWF multimer results are interpreted in conjunction with results of the initial VWD screening panel.
VWF - FVIII Binding Assay
Normally FVIII circulates bound to VWF with a half-life of 8-12h. Structurally abnormal (type 2N VWD) or absent (type 3 VWD) VWF will reduce the half-life of FVIII thereby lowering plasma concentration. Type 2N can be mistaken for mild Haemophilia A because of low FVIII levels, with treatment implications. Type 2N is characterized by a reduced VWF: FVIIIB, while this is normal in Haemophilia A.
The ELISA VWF-FVIII binding assay (VWF: FVIIIB) evaluates the capacity of VWF to bind FVIII and is only performed in cases of disproportionality between VWF and FVIII, below a FVIII: C/VWF: Ag ratio<0.5.
VWF: FVIIIB is not widely used and only performed in specialized laboratories. It provides an accurate measurement but will not be as accurate as genetic testing as patients with type 2N can be homozygous for one single 2N mutation or compound heterozygous for two 2N mutations or a single 2N that is co-inherited with a null allele. Despite this limitation, and because genetic testing is not available in all centers, the VWF: FVIIIB can be used as type 2N screening. Severely decreased VWF: FVIIIB results denotes type 2N VWD, while a moderate decrease of VWF:FVIIIB suggests heterozygous type 2N (but does not exclude Haemophilia A).
VWF Propeptide
The mature VWF and VWF propeptide stay non-covalently associated in a 1:1 ratio and are stored into the WPB (endothelial cells) and a-granules (megakaryocytes) for a regulated simultaneously release [50] into circulation. The VWF propeptide circulates independently with fixed half-life of 2–3 hours, whereas the mature VWF has a 8–12 hours half-life [51], which can vary in different VWD (sub) types. This means that VWF propeptide assay (VWFpp) is a measure for VWF synthesis/secretion, and the VWFpp/VWF: Agratio can indicate an increased VWF elimination/clearance as it is in type 1C, 2B and 2A VWD [52], either by proteolysis of VWF due to ADAMTS-13, or through removal by macrophages in liver and spleen.
In general, an increased VWFpp/VWF: Ag is associated with a shorter VWF half-life, although some patients show normal ratio’s with rapid clearance. An accurate diagnosis of patients with increased clearance, i.e. type 1C, has important implications on patient treatment. They may require VWF concentrates instead of Desmopressin, because the increase inendogenous VWF after Desmopressin infusion will dissipate quickly.
Because the VWFpp assay has become more available in the last few years, a higher incidence of increased VWF clearance within classic type 1 patients has been found. This may indicate that type 1C is more common than previously suspected. The ELISA assay is currently not routinely included in the VWD diagnostic setup but several published data suggest that the implementation would be a valuable addition to the diagnostic panel.
Genetic Testing
The current VWD diagnosis/classification [6] is not restricted to VWF gene mutations. A significant proportion of VWD patients, mainly type 1 (40% of all type 1 cases), have no identifiable mutation [42] and other genes (e.g. blood group) may contribute to the VWD phenotype. A higher linkage is seen in qualitative defects (type 2 VWD) and type 3 VWD.
Identification of causal mutations is complicated by the large size of the VWF gene, highly polymorphic, and the presence of a pseudogene located on chromosome 22q11-13. Full genetic analysis is done by DNA sequencing and Multiplex ligation-dependent probe amplification but is time consuming and therefore limited to expert labs. All identified mutations are collected in the ISTH/EAHAD VWD database. Although it contains most known VWF mutations, the VWD (sub) classification dependents on the laboratory panel results and several mutations are reported with more than one classification. As a consequence there is no gold standard in VWD classification.
Although genetic testing is not required for VWD diagnosis and classification, it can be valuable in case of the presence of a known VWF mutation in order to perform a genetic-based family study as it is not hampered by intra-individual variations of the VWF parameters.
Intra-Individual Variation
As acute phase reactant, VWF can vary importantly in the same patient at different occasions with levels influenced dramatically related to physical activity, stress, inflammation, medication and hormonal influences with values rising by a factor 2-3 [53], requiring multiple blood samplings to make the VWD diagnosis. VWF levels also increase with age [54] masking a VWD phenotype in patients who may still be at risk of bleeding.
As VWF levels are very much influenced by ABO blood groups, this patient specific factor has to be considered when interpreting the VWF results if VWD is being considered. Compared with non-O individuals, Blood group O individuals have 25% lower [55] levels, but this does not necessarily cause bleeding symptoms or VWD. Therefore it is required to use different reference ranges for blood group O and non-O next to general reference intervals.
Conclusion
Over the years remarkable advances have been made in the understanding of VWD with introduction of new technologies that can contribute to the improvement of diagnosis, classification and treatment. Although widely used, the currently available laboratory assays are not infallible; often unreliable results can lead to misdiagnosis. It is increasingly apparent that differences in results among laboratories and the use of more specialized assays have an impact on VWD diagnosis/classification. This has been demonstrated in several population studies where patients, initially classified as type 1, were reclassified as type 2 based on their VWF multimer distribution [56,57]. Laboratories are recommended to establish their own reference ranges along with strict quality control procedures. Standardization of all assays is also an ongoing effort led by the ISTH.
An algorithm to diagnose/classify VWD is also recommended by the ISTH. Because no single test gives information on both quantity and quality of VWF, it is necessary to put all the pieces of the puzzle together to obtain the whole diagnostic picture.
In most laboratories, the diagnosis is supported by a basic panel with the most common assays (VWF: Ag, VWF: GPIb and FVIII: C). All three tests are recommended for initial VWD screening and results may not only establish the diagnosis but also suggest the main type of VWD (1, 2 and 3) present.
VWF levels can vary widely by preanalytical variables and within the same individual making a correct diagnosis of (sub) types difficult, which may reflect on treatment. Therefore it is recommended to repeat tests at different times to confirm (suspected) VWD diagnosis.
The currently used automated VWF: GPIb assays, with or without ristocetin, reduce operator imprecision and workload and are more time-saving. All these assays have been shown to have a better intraand inter assay variation with an improved LLOD compared with the “gold” standard VWF: RCo technique and they also improve the accuracy with which to distinguish type 1 from type 2M even when VWF: Ag levels are relatively low.
The need for specialized assays such as VWF: MM, VWF:CB, VWF pp and VWF:FVIIIB presents another challenge in accurate VWD diagnosis.
The VWF: CB assay is consistently more sensitive to HMWM losses than VWF: GPIb. Its addition to the basic panel, allows to classify VWD according to the current ISTH/SSC classification [6] (type 1, 2A/B, 2M, 2N and 3 VWD) with both VWF GPIb/VWF: Ag and VWF:CB/VWF: Aghavinga great importance to distinguish between type 1 VWD, type 2A,B and type 2M, without distinction between 2A and 2B. The latter is usually done by the RIPA.
The VWF multimeric results are qualitatively (arbitrarily) interpreted in conjunction with results of the initial screening panel and help to determine VWD subtypes, distinguishing type 2A from 2B or subtyping 2A into 2A/IIA, IIC, IID and IIE. Therefore, the VWF: MM should only be performed if the screening panel identifies an aberrant result (e.g.low VWF: GPIb and/or VWF:CB/VWF: Ag) [58]. The semi-automated HYDRAGEL VW multimer assay can function as an initial screening of VWF multimers and is a useful addition to the VWD diagnosis in routine laboratories in conjunction with the VWD algorithm. However, it cannot fully replace the “gold “standard method since the latter shows information about triplet structure and allows modifications of the technique to “study” the underlying mechanisms.
Although genetic testing is not required for VWD diagnosis and there is no guideline under which condition genetic testing should be performed, the detection of VWF mutationsis especially important in specific situation where molecular diagnosis will help to guide treatment (type 2N and 2B) or when results are doubtful and genetic results can serve as confirmation of VWD diagnosis/classification. In case of the presence of a knownVWD mutation, a genetic-based family study can be valuable because it is not hampered by intraindividual fluctuations of VWF parameters. Normally the diagnosis of recessively inherited type 3 is unmistakable (with some exceptions) by solely measuring VWF:Ag in contrast with heterozygous type 3 mutation carriers because they mimic a type 1. Therefore genetic analysis of potential carriers within a family of a type 3 patient is justified.
A significant limitation of VWD testing is the inability to measure VWF functions under physiological flow conditions as they function in vivo. The current in vitro assays are all performed under static conditions and may not adequately reflect thrombus formation under physiological shear stress conditions.
In general, to diagnose and classify VWD in a most accurate way and with more confidence, we recommend routine laboratories, which are mostly limited in time, personal staff, expert knowledge, etc. to choose methodologies for the different assays with the best balance between accessibility and accuracy. Therefore mostly automated assays are preferred above the more manual (ELISA) methods. LIA-based or CLIA assays (unfortunately only available for use on an AcuStar®) for VWF: Ag and VWF: CB; Innovance VWF Ac (VWF: GPIbM); WB-RIPA and semi-automated VW multimer assay (if a HYDRASYS® 2 Scan is available)in conjunction with a diagnostic algorithm aids to classify VWD according to the ISTH/SSC classification with subdivision of type 2 VWD into 2A, 2B, 2M, and 2N. This panel also allows an additional subdivision of type 2A into IIA, IIC, IID and IIE, which is interesting for research purposes as it provides more information on differences in sensitivity to cleavage, dimerization and multimerization.
Acknowledgements
The author thanks A.G for his critical advice.
Author Contributions
I.V wrote the manuscript. A.G critically revised and approved the final version of the manuscript.
References
- Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987; 69: 454-9.
- Mancuso DJ, Tuley EA, Westfield LA, Shelton-Inloes BB, Sorace JM, et al. Structure of the gene for human von Willebrand factor. J Biol Chem. 1989; 264: 19514-27.
- Zhou YF, Eng ET, Zhu J, Lu C, Walz T, Springer TA. Sequence and structure relationships within von Willebrand factor. Blood. 2012; 120: 449-58.
- Sadler JE. Von Willebrand factor assembly and secretion. J Thromb Haemost. 2009; 7: 24-7.
- Lenting PJ, Neels JG, van den Berg BM, Clijsters PP, Meijerman DW, Pannekoek H, et al. The light chain of factor VIII comprises a binding site for low density lipoprotein receptor-related protein. J Biol Chem. 1999; 274: 23734-9.
- Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand factor. J Thromb Haemost. 2006; 4: 2103-14.
- Sadler JE. New concepts in von Willebrand disease. Annu Rev Med. 2005; 56: 173-91.
- Laffan MA, Lester W, O’Donnell JS, Will A, Tait RC, Goodeve A, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol. 2014; 167: 453-65.
- James PD, Connell NT, Ameer B, Di Paola J, Eikenboom J, Giraud N, et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021; 5: 280-300.
- Sadler JE. Low von Willebrand factor: sometimes a risk factor and sometimes a disease. Hematology Am Soc Hematol Educ Program. 2009: 106-12.
- Leebeek FWG, Boender J. Low VWF: an established mild bleeding disorder?. Blood. 2017; 130: 2241-2.
- Michiels JJ, Berneman Z, Gadisseur A, van der Planken M, Schroyens W, et al. Classification and characterization of hereditary types 2A(2B):2C, 2D, 2E, 2M, 2N, and 2U (unclassifiable) von Willebrand disease. Clin Appl Thromb Hemost. 2006; 12: 397-420.
- Schneppenheim R, Budde U. Von Willebrand factor: the complex molecular genetics of a multidomain and multifunctional protein. J Thromb Haemost. 2011; 9: 209-15.
- Ng C, Motto DG, Di Paola J. Diagnostic approach to von Willebrand disease. Blood. 2015; 125: 2029-37.
- Sanders YV, Groeneveld D, Meijer K, Fijnvandraat K, Cnossen MH, van der Bom JG, et al. Von Willebrand factor propeptide and the phenotypic classification of von Willebrand disease. Blood. 2015; 125: 3006-13.
- Eikenboom JC. Congenital von Willebrand disease type 3: clinical manifestations, pathophysiology and molecular biology. Best Pract Res Clin Haematol. 2001; 14: 365-79.
- Leebeek FW, Eikenboom JC. Von Willebrand’s disease. N Engl J Med. 2016; 375: 2067-80.
- Favaloro EJ, Bonar R, Marsden K. Lower limit of assay sensitivity: an underrecognised and significant problem in von Willebrand disease identification and classification. Clin Lab Sci. 2008; 21: 178-83.
- Scott JP, Montgomery RR, Retzinger GS. Dimeric Ristocetin flocculates proteins, binds to platelets, and mediates von Willebrand factor-dependent agglutination of platelets. J Biol Chem. 1991; 266: 8149-55.
- Ozarda Y, Higgins V, Adeli K. Verification of reference intervals in routine clinical laboratories: practical challenges and recommendations. Clin Chem Lab Med. 2018; 57: 30-7.
- Salem RO, Van Cott EM. A new automated screening assay for the diagnosis of von Willebrand disease. Am J Clin Pathol. 2007; 127: 730-5.
- Chen D, Tange JI, Meyers BJ, Pruthi RK, Nichols WL, Heit JA. Validation of an automated latex particle-enhanced immunoturbidimetric von Willebrand factor activity assay. J Thromb Haemost. 2011; 9: 1993-2002.
- Patzke J, Budde U, Huber A, Méndez A, Muth H, Obser T, et al. Performance evaluation and multicentre study of a von Willebrand factor activity assay based on GPIb binding in the absence of Ristocetin. Blood Coagul Fibrinolysis. 2014; 25: 860-70.
- Flood VH, Gill JC, Morateck PA, Christopherson PA, Friedman KD, Haberichter SL, et al. Gain-of-function GPIb ELISA assay for VWF activity in the Zimmerman Program for the Molecular and Clinical Biology of VWD. Blood. 2011; 117: e67-74.
- Cabrera N, Moret A, Caunedo P, Cid AR, Vila V, España F, et al. Comparison of a new chemiluminescent immunoassay for von Willebrand factor activity with the Ristocetin cofactor-induced platelet agglutination method. Haemophilia. 2013; 19: 920-5.
- Verfaillie CJ, De Witte E, Devreese KM. Validation of a new panel of automated chemiluminescence assays for von Willebrand factor antigen and activity in the screening for von Willebrand disease. Int J Lab Hematol. 2013; 35: 555-65.
- Flood VH, Gill JC, Morateck PA, Christopherson PA, Friedman KD, Haberichter SL, et al. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by Ristocetin cofactor. Blood. 2010; 116: 280-6.
- Vangenechten I, Mayger K, Smejkal P, Zapletal O, Michiels JJ, Moore GW, et al. A comparative analysis of different automated von Willebrand factor glycoprotein Ib-binding activity assays in well typed von Willebrand disease patients. J Thromb Haemost. 2018; 16: 1268-77.
- Favaloro EJ. Clinical utility of the PFA-100. Semin Thromb Hemost. 2008; 34: 709-33.
- Favaloro EJ. The utility of the PFA-100 in the identification of von Willebrand disease: a concise review. Semin Thromb Hemost. 2006; 32: 537-45.
- Favaloro EJ, Pasalic L, Curnow J. Laboratory tests used to help diagnose von Willebrand disease: an update. Pathology. 2016; 48: 303-18.
- Favaloro EJ, Dean E, Arunachalam S, Vong R, Mohammed S. Evaluating errors in the laboratory identification of von Willebrand disease using contemporary von Willebrand factor assays. Pathology. 2022; 54: 308-17.
- Turecek PL, Siekmann J, Schwarz HP. Comparative study on collagenbinding enzyme-linked immunosorbent assay and Ristocetin cofactor activity assays for detection of functional activity of von Willebrand factor. Semin Thromb Hemost. 2002; 28: 149-60.
- Favaloro EJ. Laboratory assessment as a critical component of the appropriate diagnosis and sub-classification of von Willebrand’s disease. Blood Rev. 1999; 13: 185-204.
- Flood VH, Gill JC, Christopherson PA, Wren JS, Friedman KD, Haberichter SL, et al. Comparison of type I, type III and type VI collagen binding assays in diagnosis of von Willebrand disease. J Thromb Haemost. 2012; 10: 1425-32.
- Favaloro EJ. Collagen binding assay for von Willebrand factor (VWF:CBA): detection of von Willebrands disease (VWD), and discrimination of VWD subtypes, depends on collagen source. Thromb Haemost. 2000; 83: 127-35.
- Favaloro EJ. Evaluation of commercial von Willebrand factor collagen binding assays to assist the discrimination of types 1 and 2 von Willebrand disease. Thromb Haemost. 2010; 104: 1009-21.
- Flood VH, Schlauderaff AC, Haberichter SL, Slobodianuk TL, Jacobi PM, Bellissimo DB, et al. Crucial role for the VWF A1 domain in binding to type IV collagen. Blood. 2015; 125: 2297-304.
- Favaloro EJ, Mohammed S. Evaluation of a von Willebrand factor three test panel and chemiluminescent-based assay system for identification of, and therapy monitoring in, von Willebrand disease. Thromb Res. 2016; 141: 202- 11.
- Favaloro EJ. An update on the von Willebrand factor collagen binding assay: 21 years of age and beyond adolescence but not yet a mature adult. Semin Thromb Hemost. 2007; 33: 727-44.
- Ribba AS, Loisel I, Lavergne JM, Juhan-Vague I, Obert B, Cherel G, et al. Ser968Thr mutation within the A3 domain of von Willebrand factor (VWF) in two related patients leads to a defective binding of VWF to collagen. Thromb Haemost. 2001; 86: 848-54.
- Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand disease (MCMDM-1VWD). Blood. 2007; 109: 112-21.
- James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007; 109: 145-54.
- Laffan M, Brown SA, Collins PW, Cumming AM, Hill FG, Keeling D, et al. The diagnosis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organization. Haemophilia. 2004; 10: 199-217.
- Cattaneo M, Hayward CP, Moffat KA, Pugliano MT, Liu Y, Michelson AD. Results of a worldwide survey on the assessment of platelet function by light transmission aggregometry: a report from the platelet physiology subcommittee of the SSC of the ISTH. J Thromb Haemost. 2009; 7: 1029.
- Schmidt DE, Bruzelius M, Majeed A, Odeberg J, Holmström M, ÅgrenA. Whole blood Ristocetin-activated platelet impedance aggregometry (multiplate) for the rapid detection of von Willebrand disease. Thromb Haemost. 2017; 117: 1528-33.
- Budde U, Schneppenheim R, Plendl H, Dent J, Ruggeri ZM, Zimmerman TS. Luminographic detection of von Willebrand factor multimers in agarose gels and on nitrocellulose membranes. Thromb Haemost. 1990; 63: 312-5.
- Vangenechten I, Gadisseur A. Improving diagnosis of von Willebrand disease: reference ranges for von Willebrand factor multimer distribution. Res Pract Thromb Haemost. 2020; 4: 1024-34.
- Favaloro EJ. Detailed von Willebrand factor multimer analysis in patients with von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM-1VWD): a rebuttal. J Thromb Haemost. 2008; 6: 1999-2001.
- Vischer UM, Wagner DD. Von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood. 1994; 83: 3536-44.
- van Mourik JA, Boertjes R, Huisveld IA, Fijnvandraat K, Pajkrt D, van Genderen PJ, et al. Von Willebrand factor propeptide in vascular disorders: A tool to distinguish between acute and chronic endothelial cell perturbation. Blood. 1999; 94: 179-85.
- Sztukowska M, Gallinaro L, Cattini MG, Pontara E, Sartorello F, Daidone V, et al. Von Willebrand factor propeptide makes it easy to identify the shorter von Willebrand factor survival in patients with type 1 and type Vicenza von Willebrand disease. Br J Haematol. 2008; 143: 107-14.
- Blann AD. Von Willebrand factor antigen as an acute phase reactant and marker of endothelial cell injury in connective tissue diseases: a comparison with CRP, rheumatoid factor, and erythrocyte sedimentation rate. Z Rheumatol. 1991; 50: 320-2.
- Konkle BA. Von Willebrand factor and aging. Semin Thromb Hemost. 2014; 40: 640-4.
- Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood. 1987; 69: 1691-5.
- Eikenboom J, Van Marion V, Putter H, Goodeve A, Rodeghiero F, Castaman G, et al. Linkage analysis in families diagnosed with type 1 von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 VWD. J Thromb Haemost. 2006; 4: 774-82.
- Budde U, Schneppenheim R, Eikenboom J, Goodeve A, Will K, Drewke E, et al. Detailed von Willebrand factor multimer analysis in patients with von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM- 1VWD). J Thromb Haemost. 2008; 6: 762-71.
- Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008; 14: 171-232.