
Research Article
Thromb Haemost Res 2023; 7(1): 1086.
Acute Promyelocytic Leukaemia in Adults at Chris Hani Baragwanath Academic Hospital
McMillan B, Patel M*, Rahman F, Philip V, Lakha A and Waja MF
¹McMillan B. Department of Medicine, University of the Witwatersrand, Johannesburg, South Africa
²Clinical Haematology Unit, Department of Medicine, Chris Hani Baragwanath Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg,South Africa
*Corresponding author: Patel M Emeritus Professor, Clinical Haematology Unit, Department of Medicine, Chris Hani Baragwanath Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, P O Box 96092, Brixton, 2019, Johannesburg, South Africa
Received: November 18, 2022; Accepted: January 03, 2022; Published: January 09, 2023
Abstract
Background: Acute Promyelocytic Leukaemia (APL) is a subtype of Acute Myeloid Leukaemia (AML). APL is a medical emergency due to its high early mortality rate, most commonly due to bleeding complications resulting from an associated coagulopathy. However, APL is eminently treatable.
Aim: The aim of the study was to review the clinical profile as well as the outcome of adult patients presenting with APL, to a tertiary, public sector hospital (Chris Hani Baragwanath Academic Hospital -CHBAH) in Soweto, Johannesburg, South Africa.
Patients and Methods: This study entailed a retrospective review of 79 evaluable patients with APL, seen over a 25 year period.
Results: Of the 79 evaluable patients, there were 39 males (49.37%) and 40 females (50.63%), with a female to male ratio of 1.03:1. The median age at presentation was 32 years. APL coagulopathy was evident at presentation, with 82.05% of the patients presenting with bleeding and 4.29% presenting with thrombosis. Laboratory evaluation revealed that 93.42% of the patients had anaemia and 97.37% of the patients had thrombocytopenia, while 63.93% of the patients had evidence of a DIC (disseminated intravascular coagulopathy). The Sanz risk score indicated high-risk APL in 26.32% of the patients. HIV seropositivity was present in 17.72% of the patients. After excluding patients with “early mortality’, the median survival was 43.7 months.
Conclusion: APL most commonly presents with bleeding manifestations, anaemia, thrombocytopenia and DIC, which contribute to a high “early mortality’ rate. APL is eminently treatable with appropriate supportive care and specific therapy (including ATRA – all trans retinoic acid, arsenic trioxide and chemotherapy). Therefore, early diagnosis and timeous referral of APL patients is of paramount importance in order to decrease the high early mortality and improve the overall outcome of patients harbouring the disease.
Keywords: APL; AML; ATRA; HIV; South Africa
Abbreviations: APL: Acute Promyelocytic Leukaemia; AML: Acute Myeloid Leukaemia; CHBAH: Chris Hani Baragwanath Academic Hospital; HIV: Human Immunodeficiency Virus; DIC: Disseminated Intravascular Coagulopathy; ATRA: All-Trans Retinoic Acid; ATO: Arsenic Trioxide; MCC: Medicines Control Council; SAHPRA: South African Health Products Regulatory Authority; LPA2005: Leucemia promielocitica aguda; PETHEMA: Programa De Estudio Y Tratamiento De Las Hemopatias Malignas; GIMEMA: Gruppo Italiano Malattie Ematologiche dell’ Adulto; AIDA: All-Trans Retinoic Acid and Idarubicin
Introduction
Acute Myeloid Leukaemia (AML) is a haematological malignancy that results in a clonal expansion of myeloblasts [1]. Acute Promyelocytic Leukaemia (APL) is a subtype of AML, with a characteristic genetic abnormality, t(15;17)(q22;q21) [2,3]. APL is a medical emergency, with a high early mortality rate primarily due to an associated Disseminated Intravascular Coagulopathy (DIC) and hyperfibrinolysis, with a resultant increased bleeding and thrombotic tendency [4]. As a result of the development of specific molecularly targeted therapeutic agents such as All-Trans Retinoic Acid (ATRA) and Arsenic Trioxide (ATO), as well as improved supportive care, APL is now a potentially curable disease. Cure rates in the literature are currently 75-80% [5]. APL has an unusual age-related incidence and is primarily a disease of young to middle aged individuals [6].
The aim of this study was to review the demographic and clinical profile of patients diagnosed with APL at the Clinical Haematology Unit, Department of Medicine, Chris Hani Baragwanath Academic Hospital (CHBAH) over a 25-year period, from 01-01-1994 to 31-12-2019.Particular note was made regarding early death rates, presentation immunophenotypes, management and the effects that HIV may have had on presentation and outcomes.
The findings of this study, which formed part of the Master of Medicine dissertation of Dr BP McMillan (University of the Witwatersrand, 2020) are presented [7].
Patients and Methods
This study was a retrospective review of adult patients seen at the Clinical Haematology Unit, Department of Medicine, Chris Hani Baragwanath Academic Hospital with a confirmed diagnosis of APL based on morphologic, immunophenotypic and genetic/molecular criteria, during the 25-year period 01- 01-1994 to 31-12-2019. A sample size of 79 evaluable patients was obtained.
CHBAH is a large, tertiary, public sector, teaching hospital located in Soweto, Johannesburg and is affiliated to the University of the Witwatersrand, Johannesburg, South Africa. It serves a population in excess of 1.5 million people in Soweto (and is also a tertiary referral centre for the southern part of Gauteng, the North West and Northern Cape provinces) and has approximately 3200 beds [8].
Data was obtained retrospectively from the patient files and NHLS (National Health Laboratory Services), after obtaining consent from the relevant authorities and the Human Research Ethics Committee (HREC), University of the Witwatersrand, Clearance Certificate Number: M191010.
Data collection using a questionnaire focused largely on the objectives of the study: obtaining information on the diagnosis, demographics, clinical presentation, management, complications and outcome of the patients. The relevance of the HIV association was also documented. The information was entered onto an Excel spreadsheet and analysed using the appropriate statistical tests and with the assistance of a statistician.
Results
There were 79 evaluable patients with APL included in this study during the 25-year period 01-01-1994 to 31-12-2019. There were 39 males (49.37%) and 40 females (50.63%), with a female to male ratio of 1.03:1. The median age at presentation of patients with APL was 32 years with an interquartile range of 24 – 46 years.
The APL coagulopathy was evident at presentation, with 82.05% of patients presenting with bleeding, and 4.29% with thrombosis. Most commonly, bleeding was noted at multiple (two or more) sites, in 60.76% of the patients, followed by gum bleeding in 50.63%, cutaneous bleeding in 43.04% and epistaxis in 30.38% of patients, at presentation (Table 1). In keeping with this, the most common clinical sign at presentation was pallor, being clinically apparent in 81.01% of the patients with APL (Table 2).
![]()
Site
N
%
Multiple sites (two or more sites)
48
60.76
Gums
40
50.63
Cutaneous (ecchymosis, petechiae, purpura)
34
43.04
Epistaxis
24
30.38
Venepuncture sites
21
26.58
PV bleeding
12
15.19
Buccal mucosa (petechiae, purpura, haemorrhagic bullae)
9
11.39
Haemoptysis
6
7.59
GI tract bleeding (melaena, haematemesis, haematochezia)
5
6.33
Haematuria
5
6.33
Ocular
3
3.80
Post-surgery (LLETZ, circumcision, biopsy)
3
3.80
Penile
2
2.53
Intracranial
1
1.27
APL: acute promyelocytic leukaemia
PV: per vaginal
GI: gastrointestinal
LLETZ: large loop excision of the transformation zone
Table 1: Site of bleeding in patients with APL.
![]()
Sign
N
%
Pallor
64
81.01
Lymphadenopathy
23
29.11
Hepatomegaly
17
21.52
Splenomegaly
4
5.06
Jaundice
3
3.80
APL: acute promyelocytic leukaemia
Table 2: Clinical signs in patients with APL.
Laboratory evaluation revealed that 93.42% of patients had an anaemia (haemoglobin < 11g/dl) and 97.37% had clinical thrombocytopenia (platelets < 100 x 109/l). There was evidence of a DIC in 63.93% of patients. There was rapid improvement of all these parameters following initiation of therapy (Table 3). At presentation, the median haemoglobin was 6.55 g/dl, the median white cell count was 3.51 x 109/l and the median platelet count was 24 x 109/l. Other relevant blood parameters are shown in (Table 3). The Sanz risk score (which is based on the white cell and platelet counts at presentation) was used to assess risk, and 26.32% of patients had a high-risk APL (Table 4).
![]()
Median (Range)
Parameter
At Presentation
10 days post treatment
After first cycle of treatment
First clinic visit
At completion of chemotherapy
At last date seen / last visit
White cell count x 109/l
3.51 (0.51-144)
2.04(0.46-26.9)
5.06 (1.77-24.08)
5.39 (2.3-11.72)
5.03 (3.42-7.8)
5.12 (0.19-203)
Haemoglobin g/dl
6.55 (3.1-13.6)
8.1 (6.1-10.7)
9.65 (7.01-14)
11.9 (6.5-15.8)
12.85 (6.5-15.7)
10.8 (3.1-16.5)
Haematocrit l/L
0.19 (0.10-0.37)
0.25 (0.19-0.34)
0.31 (0.23-0.42)
0.36 (0.27-0.43)
0.36 (0.27-0.45)
0.26 (0.10-0.41)
MCV fl
90 (73.3-109)
86.6 (78-97.5)
91.2 (82-104.1)
90.4 (82.1-103.1)
90.35 (83.8-98.8)
88.0 (74.1-111)
Platelets x 109/L
24 (2-178)
39.5 (10-225)
430 (42-821)
356.5 (87-987)
270.5 (87-467)
154.5 (2-512)
Na mmol/L
137 (124-154)
137 (122-179)
139 (132-148)
139 (138-142)
141 (141-141)
137 (128-172)
K mmol/L
3.7 (2.5-4.9)
4.1 (2.5-5.1)
4.1 (2.8-5.3)
4 (3.1-4.3)
4.5 (4.5-4.5)
3.5 (2.5-5.5)
Cl mmol/L
100 (89-111)
102 (84-135)
102 (89-111)
100.5 (99-102)
100 (100-100)
104 (89-133)
HCO3 mmol/L
19 (1.4-31)
18 (1.1-31)
21 (2-30)
25 (4.6-25)
22 (22-22)
20 (2.8-28)
Urea mmol/L
6.1 (1.5-40.3
6.2 (1.9-22.2)
4.7 (1.7-96)
4.65 (3.4-50)
4 (4-4)
6.9 (2.4-38)
Creatinine umol/L
75(45-450)
66 (40-149)
61 (38-105)
67 (56-84)
69 (69-69)
80 (36-399)
UA mmol/L
0.32 (0.08-0.8)
0.32 (0.32-0.32)
0.21 (0.19-0.33)
-
-
-
LDH mmol/L
601.5 (203-4506)
1010 (1010-1010)
620.5 (235-1006)
406 (406-406)
-
-
INR
1.3 (0.87-2.85)
1.21 (0.88-1.64)
1.08 (0.85-1.76)
-
-
1.39 (0.91-6.2)
Fibrinogen g/L
2.1 (0.5-21)
3.65 (1.1-6.8)
3.4 (1.9-8.5)
-
-
3.5 (2.03-40)
PTT sec
32.7 (20.7-60)
32 (20-59.5)
34.5 (27.8-64)
-
-
39.1 (11.5-60.8)
APL: acute promyelocytic leukaemia
MCV: mean corpuscular volume
UA: uric acid
LDH: lactate dehydrogegnase
INR: international normalised ratio
PTT: partial thromboplastin time
Table 3: Laboratory findings in patients with APL.
![]()
Sanz group
N
%
Low risk
11
14.47
Intermediate risk
45
59.21
High risk
20
26.32
Total
76
100
APL: acute promyelocytic leukaemia
Table 4: Sanz score risk stratification of patients with APL at presentation.
HIV was the most common co-existing comorbidity in this study, being present in 17.72% of all patients (14 patients).
When bone marrow morphology was assessed, 67 patients (91.78%) had the typical, hypergranular morphology, and six patients had a hypogranular variant. The characteristic t (15;17) was evident in 49 of the 51 patients (96%) that were tested (bearing in mind that in the early part of the study, the test was not routinely available).
Management involved supportive therapy with blood and blood products, and specific therapy, most commonly with ATRA and daunorubicin during induction and consolidation. Maintenance therapy included ATRA, methotrexate and mercaptopurine. There were 21 patients who received re-induction therapy, five of whom were for failed induction and the remaining patients for relapsed disease. Early complications, defined as complications occurring within the first 30 days from initia-tion of therapy, were found in 89.33% of patients, and 83.54% of these were bleeding complications (Table 5). Thirty-seven patients achieved remission following induction therapy.
![]()
Early complications
N
%
Haemorrhage
66
83.54
Infection
29
36.71
Renal failure
21
26.58
Differentiation syndrome
8
10.13
TED
6
7.59
Other
6
7.59
Late complications
Infection
18
22.78
Renal failure
8
10.13
Other
5
6.33
TED
2
2.53
Differentiation syndrome
1
1.27
APL: acute promyelocytic leukaemia
TED: thromboembolic disease
Table 5: Complications in patients with APL.
At the last date seen, 39.24% of patients were in remission, 49.37% of patients had died and 3.8% had relapsed disease. Overall, the median survival was 7.8 months, which improved to 43.7 months once early mortality was excluded (Figure 1). There was no statistically significant difference in the survival time between HIV seropositive and seronegative patients. There was also no statistically significant difference in survival time across the low, intermediate and high-risk groups as calculated by the Sanz score.
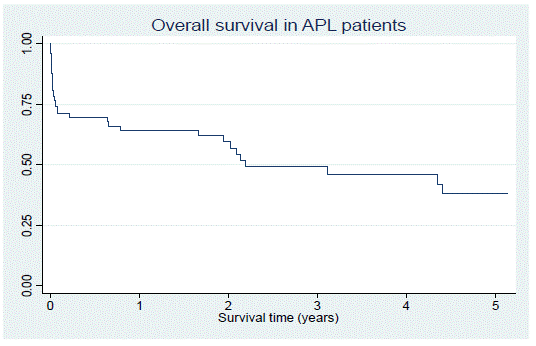
Figure 1: Overall survival in APL patients.
Discussion
AML is the most common acute leukaemia in adults. APL is a subtype of AML. Both AML and APL have a younger age of incidence in African studies which is reflected in the findings of this study [6,9,10]. The median age at presentation of patients with APL in this study was 32 years with an interquartile range of 24 – 46 years.
HIV was the most common co-existing comorbidity in this study, being present in 17.72% of all patients. It is not clear if there is a causative link or an association between HIV and APL, or if this is simply an indication of the high prevalence of HIV in this particular age group, in which APL commonly occurs. There was a clear increase in the number of APL patients with HIV from the first to the second half of the study, however the number of patients diagnosed with APL had also increased over this timeframe. The higher HIV association in the latter half of the study is also likely to be a reflection of the ongoing high burden of HIV seropositivity in the South African context [11]. Importantly, the clinical presentation, management and outcome were similar in both HIV seronegative and HIV seropositive patients with APL, in this study.
The most common symptom at presentation in these patients was bleeding, found in 82.05% of APL patients. This is in keeping with the APL coagulopathy, including DIC, which is commonly found at presentation [4].
High-risk APL, as classified by the Sanz score, includes patients with a WCC of > 10 x 109/l. In the LPA99 and LPA2005 trials, 25% and 29% of patients, respectively, had a high-risk APL, which is similar to the percentage seen in our study (26.32%) [4,12,13].
This study was performed over a long time period, and the diagnosis of APL was made based on methodology available at the given time. Initially, the diagnosis was made clinically, morphologically and with the use of immunohistochemistry. Later, flow cytometry and cytogenetics were available and these methods were also used. Finally, the diagnosis was made molecularly, in addition to the other diagnostic tools. Towards the latter part of the study, the diagnosis was more comprehensive, using both molecular and cytogenetic techniques. When bone marrow morphology was assessed, 67 patients (91.78%) had the typical hypergranular morphology. Six patients had a hypogranular variant, which is associated with a worse prognosis. However 4 of these 6 these patients (66.6%) achieved remission post induction.
The immunophenotype in our high-risk population of APL patients as defined by the Sanz score was similar to that seen in the general population of APL patients. However, these highrisk patients had a higher percentage of CD2 positivity at 30%, CD34 positivity at 25% and CD56 positivity at 5%, which is in keeping with the worse prognosis predicted by both the immunophenotype and the high-risk nature of the disease conferred by the high WCC [14].
There was a t(15;17) in 49 of the 51 patients (96%), that were tested. This is associated with a good prognosis due to a good response to ATRA [15]. Additionally, when molecular testing became available, there were 38/38 patients (100%), who tested positive for PML-RARA fusion gene [15].
There were delays in both the appropriate referral of patients to our facility, as well as in the initiation of treatment. The majority of patients diagnosed with APL were either referred to our facility immediately (80.77%) or the following day (12.82%), with the remaining 6.4% of patients experiencing a delay of between 2 and 15 days. Once patients were referred to the Clinical Haematology Unit, treatment was initiated in 51.38% of patients on the same day, and 16.67% the following day, with the remaining patients (31.95%), experiencing delays of up to 19 days. This could be accounted for by concomitant comorbidities precluding immediate treatment, patient’s refusing therapy, or other barriers to treatment such as medication stock outs (with particular reference to ATRA).
Supportive therapy is a cornerstone of treatment in acute leukaemia, and was offered to all the patients who were treated. In particular, this included the management of the coagulopathy with the use of blood and blood products, and initiation of ATRA orally, once the diagnosis of APL was suspected.
The standard of therapy for APL during induction currently includes the use of ATRA, an anthracycline and ATO in different combinations [3]. In our study, it was found that 91.14% of patients received ATRA (data was missing for three patients). Induction therapy also included the use of daunorubicin in 82.61% of patients, and 20.29% received cytarabine as well. During the study period ATO was not readily available in the South African public sector for induction therapy in APL. Although it is not clear which anthracycline may be best for induction, there is some evidence to suggest that idarubicin may result in improved survival in comparison to daunorubicin [6,13].
Consolidation therapy in APL is recommended to be given in three cycles of monthly therapy once remission has been achieved following induction, as per the LPA2005 trial [4]. This trial used ATRA with an anthracycline, and high-risk patients were also given cytarabine [4]. The most commonly received regimen consisted of ATRA and daunorubicin, consistent with this evidence. A large randomised control trial also showed benefit in survival with the use of ATO combined with ATRA and daunorubicin for consolidation therapy, however, ATO was only available in South Africa on a named-patient basis through approval by the MCC / SAHPRA during the study period [6].
There were 23 patients in our study who were documented to have received maintenance therapy, which consisted of ATRA, methotrexate and mercaptopurine, consistent with the regimen in the PETHEMA (Programa de Estudio y Tratamiento de las Hemopatias Malignas) trial [13]. The GIMEMA (Gruppo Italiano Malattie Ematologiche dell’ Adulto) group in the AIDA- 0493 and AIDA-2000 (All-trans retinoic acid and idarubicin) trials found that patients receiving ATRA as part of maintenance therapy had better outcomes and fewer relapses [17].
A total of 37 patients achieved remission following induction, and 21 patients were in remission post consolidation. Thirty-one patients were in remission at the last date they were seen at the Clinical Haematology Unit.
Immediate mortality (death in the first three days of treatment being initiated) occurred in twelve patients (15.18%) and was again most commonly caused by bleeding, but early mortality (death occurring more than three days after treatment initiation), which occurred in six patients (7.59%), was more likely to be the result of infection, as was seen in late mortality (more than thirty days after treatment initiation), which occurred in 15 patients (18.98%). This is consistent with what is found in the literature, with immediate mortality likely to be caused by bleeding and early mortality likely to be due to infection or the differentiation syndrome [14].
In our study, overall survival at three years was close to 50%, which is inferior to that described in the local and international literature. In a study performed in Cape Town, the overall survival at three years was 76.5% [19]. In a Tunisian study, there was a five-year overall survival of 78% [5]. In the LPA2005 study, overall survival of the patients at three years was 89%, and 88% at four years, respectively [12]. There are a number of reasons that could explain the inferior survival in our study cohort, including the high early mortality (removal of which demonstrated a much higher survival time) and barriers to receiving timeous treatment and to ongoing access to healthcare specific to our population. Although survival time appeared shorter in the HIV positive population in this study, the difference was not statistically significant. The differences in survival time in the different risk groups of APL was also not statistically significant.
Conclusion
APL, a subtype of AML, most commonly presents with bleeding manifestations, anaemia, thrombocytopenia and DIC; manifestations of the APL coagulopathy. High-risk APL (based on the Sanz score and a high WCC) was evident in 26.32% of the patients in this study. There were early complications in 89.33% of patients, 83.54% of which were the result of bleeding. This high morbidity rate contributed to the immediate and early mortality rate of 22.77% of the patients, in this study.
HIV was the most common comorbidity found in this study, and is highly prevalent in South Africa. There was no overt relationship between these two disease entities, and the association is therefore likely coincidental, with both these diseases occurring characteristically in the young to middle age groups. Further studies will be needed to better characterise this relationship.
APL is an eminently treatable disease with appropriate supportive care and specific therapy. Therefore, recognition and awareness of this entity, early diagnosis and timeous referral is of paramount importance in order to decrease the high early mortality of APL and to increase the long term outcome of patients with the disease.
References
- O’donnell M, Tallman M, Abboud C, Altman J, Appelbaum F, Arber D, et al. Acute myeloid leukaemia, version 3.2017. JNCCN. 2017; 15: 926-957.
- Blum W and Bloomfield C. Harrison’s Principles of Internal Medicine. 20th ed. Jameson J, Kasper D, Longo D, Fauci A, Hauser S, Loscalzo J, et al., editors. New York: McGraw Hill; 2018.
- Zhang Y, Wu S, Luo D, Zhou J, Li D. Addition of arsenic trioxide into induction regimens could not accelerate recovery of abnormality of coagulation and fibrinolysis in patients with acute promyelocytic leukemia. PLOS One. 2016; 11: e0147545.
- Sanz M and Montesinos P. Advances in the management of coagulopathy in acute promyelocytic leukemia. Thrombosis Research. 2020: S63-S67.
- Jeddi R, Ghedira H, Amor R, Abdennebi Y, Karima K, Mohamed Z, et al. Treatment of Acute Promyelocytic Leukemia with AIDA Based Regimen. Update of a Tunisian Single Center Study. Mediterranean Journal of Hematology and Infectious Diseases. 2011; 3: e2011033.
- Sanz M, Grimwade D, Tallman M, Lowenberg B, Fenaux P, Estey E, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European Leukemia Net. Blood. 2009; 113: 1875-1891.
- McMillan BP. Acute promyelocytic leukaemia in adults at Chris Hani Baragwanath Academic Hospital. Research report in partial fulfilment of the degree of Master of Medicine (Internal Medicine). University of the Witwatersrand, 2022.
- Research Unit. Overview of Chris Hani Baragwanath Academic Hospital. Parliament of the Republic of South Africa. 27 March 2022. [Available from: www.parliament.gov.za].
- Mokgoko D. Acute Myeloid Leukaemia and Human Immunodeficiency Virus Infection at Chris Hani Baragwanath Academic Hospital. MMed Dissertation submitted to the University of the Witwatersrand. 2018.
- Marshall R, Tlagadi A, Bronze M, Kana V, Naidoo S, Wiggill T, et al. Lower frequency of NPM1 and FLT3-ITD mutations in a South African adult de novo AML cohort. International Journal of Laboratory Hematology. 2014; 36: 656-664.
- Statistics South Africa (Stats SA), 2021.
- Sanz M, Montesinos P, Rayon C, Holowiecka A, Serna JDL, Milone G, et al. Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome. Blood. 2010; 115: 5137-5146.
- Sanz M, Martin G, Gonzalez M, Leon A, Rayon C, Rivas C, et al. Risk-adapted treatment of acute promyelocytic leukemia with all-trans-retinoic acid and anthracycline monochemotherapy: a multicenter study by the PETHEMA group. Blood. 2003; 103: 1237-1243.
- Xu F, Yin C, Wang C, Jiang X, Jiang L, Wang Z, et al. Immunophenotypes and immune markers associated with acute promyelocytic leukemia prognosis. Disease Markers. 2014; 2014: 421906.
- Grimwade D, Biondi A, Mozziconacci M, Hagemeijer A, Berger R, Neat M, et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Blood. 2000; 96: 1297-1308.
- Fan Y, Cao Y, Bai X and Zhuang W. The clinical significance of FLT3 ITD mutation on the prognosis of adult acute promyelocytic leukemia. Hematology. 2017; 23: 379-384.
- Lo-Coco F, Avvisati G, Vignetti M, Breccia M, Gallo E, Rambaldi A, et al. Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood. 2010; 116: 3171-3179.
- Xu F, Wang C, Yin C, Jiang X, Jiang L, Wang Z, et al. Analysis of early death in newly diagnosed acute promyelocytic leukemia patients. Medicine. 2017; 96: e9324.
- Shein R, Mashigo N, Toit CD, Oosthuizen J, Seftel M, Louw V, et al. Outcomes for Patients with Acute Promyelocytic Leukemia in South Africa. Clinical Lymphoma, Myeloma and Leukemia. 2021; 21: 348-352.