
Research Article
Ann Transl Med Epidemiol. 2016; 3(1): 1009.
Conjugation of Cholesterol to HIV-1 Fusion Peptide CP32M Improves its Antiviral Potency and Retainment at the Host Cell Membrane
Xiaoqian Zhang¹, Jian Hu1,2, Jie Li1,3 and Ying-Jie Wang1*
¹State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China
²Chengkou Agriculture Commission of Chongqing City, Chongqing 405900, China
3Department of Infectious Diseases, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou 325027, China
*Corresponding author: Ying-Jie Wang, State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, First Affiliated Hospital, School of Medicine, Zhejiang University, 79 Qing Chun Road, Hangzhou 310003, China
Received: June 10, 2016; Accepted: July 11, 2016; Published: July 14, 2016
Abstract
Peptides derived from the C-terminal heptad repeat (CHR) of the HIV-1 gp41 can effectively block viral entry by binding to the trimeric viral N-terminal heptad repeat (NHR) coiled-coil core. CP32M, a peptide modified from the CHR peptide CP621-652, has high antiviral potency, broad antiviral spectrum, and superior antiviral profile against CHR-resistant HIV-1 variants. However, peptide inhibitors including CP32M are expected to reach their target sites by simple diffusion in vivo which is rather inefficient, and the majority of the administered peptides will be metabolized and nullified. To overcome the problem, in this study, we designed a peptide called “CP32M-12aa-chol” in which the carboxylterminus of CP32M is extended with a flexible 12 amino acid (aa) linker followed by a cysteine residue where a cholesterol group is directly conjugated. Compared with its parental peptides that contain no cholesterol conjugation, the CP32M-12aa-cholpeptide had significantly enhanced antiviral potency but no appreciable cytotoxicity. Consistent with its anticipated mechanism of action, the antiviral potency of CP32M-12aa-chol was only reduced by 28-fold when it was pre-incubated with the host cells followed by thorough washes while under the same conditions all other tested peptides with no cholesterol conjugation lost their antiviral potencies by 159- to 3816-fold. Thus, adding a cholesterol group to the carboxyl-terminus of CP32M is likely to facilitate its targeting to and retention at the host cell membrane where the cell membrane/viral membrane fusion-mediated HIV-1 entry occurs, and such a strategy holds promise for further developing highly efficacious yet safe drugs against HIV-1.
Keywords: HIV-1; Fusion peptide; CP32M; Cholesterol; Cell membrane
Introduction
Numerous studies over the past two decades have provided definitive evidence that the gp41 “prehairpin intermediate” is an effective target for peptide- and neutralizing antibody-based fusion inhibitors to block HIV-1 viral entry [1]. Peptide-based fusion inhibitors mainly include the Class 1 inhibitors that target the NHR trimeric coiled-coil core and the Class 2 inhibitors that bind to the CHR region [2,3]. The best-characterized example of the Class 1 inhibitors is the CHR peptide T20 (residues 638-673 of gp41), also known as DP178 or enfuvirtide [4]. Unfortunately, the clinical application of T20 is limited because of its relatively low potency, low genetic barrier to drug resistance and short half-life. Subsequently developed CHR peptide C34 (residues 628-661 of gp41) or CP621- 652 (residues 621-652 of gp41) which included the pocket-binding domain (PBD) only or PBD plus the “QIWNNMT” motif (Figure 1A), respectively, exhibited more potent HIV-1 fusion inhibitory activity than T20 [2,3]. The “QIWNNMT” motif and in particular the M-T hook-like structure could stabilize the interaction between NHR and CHR in the 6-HB core or between the CHR peptide and the deep pocket on the NHR trimer and thus increasing the anti-HIV-1 activity of CHR peptides [5]. However, CHR peptides with the wild type HIV-1 gp41 sequences all tend to develop viral drug-resistance in a relatively short time.
To further improve the drug-resistant profiles and the pharmacokinetics of the CHR peptides, He et al. took the CP621–652 as a template and mutated 11 of 32 residues that are not essential for the activity or stability of the peptide, resulting in a peptide termed CP32M (Figure 1B) [6]. CP32M is highly effective against a panel of primary HIV-1 strains with distinct genotypes (group M, subtypes A–G) and phenotypes (R5 and R5X4), and is exceptionally potent against HIV-1 variants resistant to T20 and other CHR peptides including C34. Although its parental peptide CP621–652 is also potent against the drug-resistant HIV-1 viruses, CP32M exhibited much improved antiviral potencies against some T20-resistant HIV- 1 variants, with IC50s even in the picomolar range.
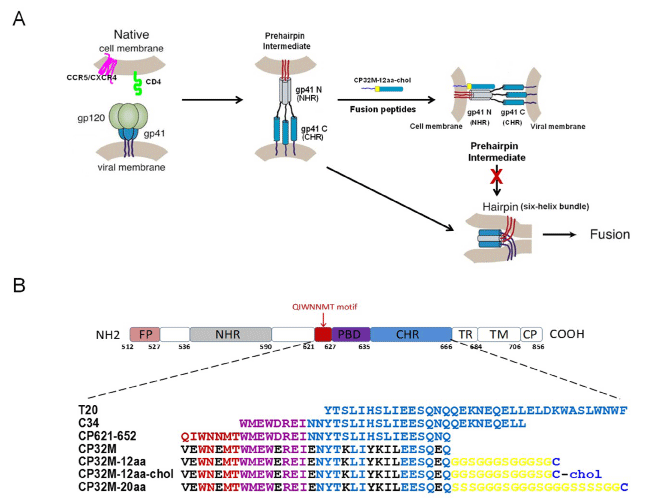
Figure 1: 1: Mechanism of action of CHR-derived peptide inhibitors and structure of CP32M-12aa-chol. (A) A model of HIV-1 gp41-mediated viral and host cellular
membrane fusion and the putative targeting sites for fusion inhibitors used in this study. The gp120 subunit of the trimeric envelope glycoprotein binds to the CD4
receptor and the chemokine coreceptor (CXCR4 or CCR5) triggering a conformational change in the gp41 subunit and causing its N-terminal heptad repeat (NHR,
“gp41 N”) and C-terminal heptad repeat (CHR, “gp41 C”) to form the so-called ��prehairpin intermediate” structure, which is further transformed into a “six-helix
bundle” structure that drives viral–cell membrane fusion. Since the “prehairpin intermediate” state lasts for approximately 15–20 min, CHR-based fusion peptides
such as CP32M-12aa-chol can bind to the exposed trimeric NHR, hampering the formation of the “six-helix bundle” and ultimately the viral entry. The CP32M-
12aa-chol anchored into the cell membrane facilitates its interaction with the viral trimeric NHR, and the 12aa Gly-Ser linker allows for fine adjustment of the
orientation and position of the CP32M portion to ensure its optimal interaction with the viral NHR trimer. (B) Schematic representation of key elements of HIV-1
gp41 and the amino acid sequences for all the peptides used in this study. The peptides T20 and C34 correspond to aa 638–673 and aa 628–661 at the CHR of
gp41, respectively. CP621-652 and CP32M originate from aa 621-652 with the latter peptide having 11 residues mutated (marked in black). The Gly-Ser linkers
are marked in yellow, the Cys residues on which cholesterol are directly conjugated are marked in blue. FP, fusion peptide; PBD, pocket-binding domain; TR,
tryptophan-rich domain; TM, transmembrane domain; CP, cytoplasmic domain.
However, after being administered into human body, peptide inhibitors including CP32M are expected to reach their target sites by simple diffusion which is rather inefficient, and the majority of the administered peptides will be metabolized and nullified. To overcome this problem, in Gallinella et al. took an innovative approach by adding a cholesterol group to the C-terminal end of the C34 peptide thereby targeting it to the host cell membrane where fusion occurs [7]. Such approach led to dramatic enrichment and sustainment of the peptide inhibitor at the site of HIV-1 entry, greatly reducing the CHR peptide concentration that is required for blocking viral entry [7]. Thus, in current study, we aimed at adding a cholesterol group to the C-terminus of the CP32M peptide to allow for its targeting to and retention at the host cell membrane and thereby further improving its antiviral potency.
Materials and Methods
In order to ensure a flexible link between the CP32M and the cholesterol group so that the CP32M peptide may have high enough chances to approach and bind to its target (i.e., the trimeric NHR coiled-coil core) when its C-terminal end is anchored into host cell membrane via the cholesterol group, we placed a 12aa- or 20aa-Gly- Ser-linker right after the C-terminus of CP32M but prior to the last Cys residue where the cholesterol group is directly attached (Figure 1B). Such Gly-rich linkers are proposed to form a flexible hinge to allow the attached peptides to extend and rotate [8]. Five peptides (C34, CP32M, CP32M-12aa, CP32M-20aa and CP32M-12aa-chol, Figure 1B for detailed sequences) were all synthesized by Chinese Peptide Company (Hangzhou, China). The purities of the peptides were approximately 95% as determined by RP-HPLC. The inhibitory effects of fusion peptides on HIV-1 infectivity were evaluated by using an enhanced green fluorescent protein (EGFP) reporter-based JLTRG cell system [9] as described previously [10]. Since the Jurkat T cell-derived JLTRG cells (which express CD4 and CXCR4) harbor a stably integrated EGFP gene driven by an HIV-1 long terminal repeat (LTR) promoter, the levels of viral infection and propagation can be determined by flow cytometry using EGFP expression as a readout [9,10].
Results
With the above HIV-1 infectivity assay system, the antiviral potencies of the five chemically synthesized CHR-based fusion peptides (C34, CP32M, CP32M-20aa, CP32M-12aa, CP32M-12aachol) were determined (Figure 2, A1-E1, Table 1). Compared with C34, CP32M had a 5.4-fold reduction in antiviral potency. Adding a 12 aa- or 20 aa-Gly-Ser-linker to the C-terminus of CP32M further reduced its antiviral potency to a small extent. Remarkably, conjugation of the cholesterol group enhanced the antiviral potency of the CP32M-12aa peptide by 6.1-fold (CP32M-12aa IC50=15.2 nM vs. CP32M-12aa-chol IC50=2.5 nM).
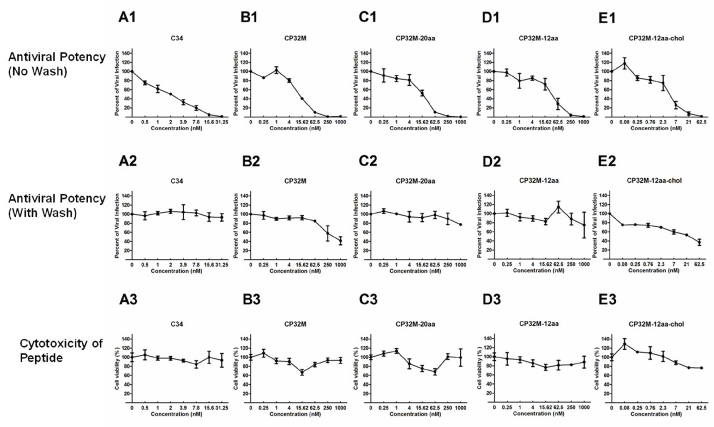
Figure 2: Antiviral potencies and cytotoxicities of the tested HIV-1 fusion peptides. (A1-E1) JLTRG cells maintained in RPMI 1640 supplemented with 1% penicillin/
streptomycin and 10% heat-inactivated fetal bovine serum, were seeded in 12-well plates at 105 cells per well, and incubated in the absence or presence of
different concentrations of the CHR peptides at 37°C in 5% CO2. After 2h, 5 x 103 of H9 cells which could release the HIVHTLV-IIIB virions were added into each
well to initiate infection. After further incubation at 37°C in 5% CO2 for 96h, the EGFP expression level indicative of viral infection was measured by flow cytometry.
Data were collected and IC50 values were calculated by SPSS20.0. The results were shown as means ± SEM from three independent experiments. (A2-E2) Each
of the tested peptide was pre-incubated with JLTRG cells at 37 °C for 2h, followed by three washes with culture medium to remove unbound peptides (no wash as
a control) and addition of H9 cells to initiate infection. After 96h, the antiviral activities of the residual peptides surviving from the washing steps were determined
by measuring the EGFP expression as described above. The results were shown as means ± SEM from three independent experiments. (A3-E3) 50 μL of each
serial diluted CHR peptide were added into equal volume of cells (3 x 103/mL per well) in 96-well plates, followed by incubation at 37°C for 96h. 10 μL of WST-1
reagent were then added into each well and the absorbance was measured 4h later at 450 nm using Model 680 Microplate Reader. The results were shown as
means ± SEM from three independent experiments.
![]()
IC50, nM
No wash
With wash
C34
CP32M
CP32M-20aa
CP32M-12aa
CP32M-12aa-chol
1.5 ± 0.2
8.1 ± 0.7
10.1 ± 3.3
15.2 ± 4.4
2.5 ± 0.5
5724.1 ± 111.9 (3816)
2489.1 ± 539.6 (307)
3622.8 ± 346.4 (359)
2420.7 ± 630.0 (159)
68.9± 13.4 (28)
The antiviral potencies of the fusion peptides without or with thorough washes were determined with EGFP reporter-based JLTRG cells infected with HIVHTLV-IIIB virions. All data were from three independent experiments and expressed as mean ± SEM. Values in parenthesis indicate relative changes (n-fold) in the IC50s of the peptides with wash compared to those with no wash.
Table 1: Antiviral potencies of cholesterol-conjugated CP32M and related peptides.
To evaluate the retention of the above tested peptides on host cell membrane, each peptide was pre-incubated with JLTRG cells at 37°C for 2h, followed by three washes with culture medium to remove unbound peptides (no wash as a control) and addition of H9 cells (that could release the HIVHTLV-IIIB virions) to initiate infection. After 96h, the antiviral activities of the residual peptides surviving from the washing steps were determined by measuring the EGFP expression as described above. Among the five peptides tested, the washing step reduced the antiviral potency of C34 to the greatest extent (by 3816-fold), followed by that of CP32M-20aa, CP32M, CP32M-12aa, and CP32M-12aa-chol (Figure 2, A2-E2, Table 1). Remarkably, with wash, the fold reduction in antiviral potency for CP32M-12aa-chol was one or two orders of magnitude lower than for all the other tested peptides that do not have the cholesterol group (Table 1), strongly indicating that cholesterol conjugation dramatically promoted the retention of the CP32M fusion peptide at the host cell membrane where the HIV-1 entry occurs. Interestingly, compared with C34, all CP32M-based peptides had much less loss of antiviral potencies upon wash (Table 1). At the tested concentrations, none of the peptides exhibited obvious cytotoxicity (Figure 2, A3-E3).
Conclusion
Taken together, in this study, we conjugated a cholesterol group to the C-terminal end of CP32M peptide and a Gly- and Ser-rich linker was also introduced to allow for flexible connection between CP32M and the cholesterol group. In a cell-based HIV-1 infectivity assay, the resulting CP32M-12aa-cholesterol peptide had significantly improved antiviral potency and much prolonged retainment at the host cell membrane over its non-conjugated parental peptides. Future studies will be directed towards testing this promising anti- HIV peptide in in vivo settings represented by appropriate animal models for HIV-1 infection.
Acknowledgments
JLTRG cells were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (catalog no. 11587), from Dr. Olaf Kutsch. This work was fanatically supported by Zhejiang Provincial Natural Science Foundation of China for Distinguished Young Scholars (Grant No. R2100226).
References
- Yi HA, Fochtman BC, Rizzo RC, Jacobs A. Inhibition of HIV Entry by Targeting the Envelope Transmembrane Subunit gp41. Curr HIV Res. 2016; 14: 283-294.
- Zhang D, Li W, Jiang S. Peptide fusion inhibitors targeting the HIV-1 gp41: a patent review (2009 - 2014). Expert Opin Ther Pat. 2015; 25: 159-173.
- Fumakia M, Yang S, Gu J, Ho EA. Protein/peptide-based entry/fusion inhibitors as anti-HIV therapies: challenges and future direction. Rev Med Virol. 2015.
- Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov. 2004; 3: 215-225.
- He Y, Cheng J, Li J, et al. Identification of a critical motif for the human immunodeficiency virus type 1 (HIV-1) gp41 core structure: implications for designing novel anti-HIV fusion inhibitors. J Virol. 2008; 82: 6349-6358.
- He Y, Cheng J, Lu H, et al. Potent HIV fusion inhibitors against Enfuvirtideresistant HIV-1 strains. Proc Natl Acad Sci U S A. 2008; 105: 16332-16337.
- Ingallinella P, Bianchi E, Ladwa NA, et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A. 2009; 106: 5801-5806.
- Reddy Chichili VP, Kumar V, Sivaraman J. Linkers in the structural biology of protein-protein interactions. Protein Sci. 2013; 22: 153-167.
- Ochsenbauer-Jambor C, Jones J, Heil M, Zammit KP, Kutsch O. T-cell line for HIV drug screening using EGFP as a quantitative marker of HIV-1 replication. Biotechniques. 2006; 40: 91-100.
- Wang A, Chen F, Wang Y, et al. Enhancement of antiviral activity of human alpha-defensin 5 against herpes simplex virus 2 by arginine mutagenesis at adaptive evolution sites. J Virol. 2013; 87: 2835-2845.