
Research Article
Austin J Trauma Treat. 2021; 6(1): 1015.
Primary Osseous Spine Tumors in Adults: A Review and Update on Diagnosis and Treatment
Emerson BD¹*, Ameri BJ¹, Downing MA² and Temple HT³
¹Broward Health Medical Center, Fort Lauderdale, Florida
²Nova Southeastern University, Davie, Florida
³Mercy Hospital, Miami, Florida
*Corresponding author: Blaze D Emerson, Broward Health Medical Center, 1600 S Andrews Ave, Fort Lauderdale, Florida
Received: April 23, 2021; Accepted: May 17, 2021; Published: May 24, 2021
Abstract
Objective: This article aims to provide a convenient and comprehensive review describing unique features of the most common primary osseous spine tumors, as well as current diagnostic and therapeutic modalities for each tumor.
Background: Primary osseous spine tumors are a rare and diverse group of neoplasms with varying biologic behavior. Clinical, radiographic, and pathologic correlation is critical in making a correct diagnosis. Prompt treatment is necessary to optimize clinical outcomes, and is based on tumor type, location, and disease stage. Most patients with spinal tumors present with a history of pain often similar in quality and intensity as non-tumoral etiologies of back pain. Spinal neoplasms, especially malignant tumors, require a multidisciplinary approach and are best treated in dedicated cancer centers to mitigate incorrect diagnoses and inappropriate treatment.
Methods: A literature review was conducted using PubMed and EBSCO. Multiple search queries for relevant articles between 2014 to present were included. Preference was given to recent articles with clinical evidence, current treatment, diagnostic modalities and/or future potential therapies and diagnostic strategies.
Results: Numerous modalities including surgery, chemotherapy, evolving immunologic and targeted therapies as well as stereotactic external beam radiation therapy are utilized to optimize care. Still, current therapeutic strategies result in significant morbidity and mortality and local disease recurrence and systemic relapse are common despite chemotherapy and advanced surgical techniques.
Conclusion: Because primary spinal tumors are uncommon, level I and II data are scarce though novel treatment strategies are emerging. Medical and orthopaedic oncologists and spine surgeons therefore should have a fundamental knowledge of the current state of literature pertaining to this topic.
Keywords: Spine; Tumor; Orthopaedics; Surgery; Chordoma; Chondrosarcoma; Multiple Myeloma; Giant Cell Tumor; Osteosarcoma; Orthopaedic Oncology; Hemangioma
Introduction
Primary osseous spine tumors are rare and represent a diverse group of neoplasms with varying biologic behavior. Clinical, radiographic, and pathologic correlation is critical in making a correct diagnosis. The tumor type can often be predicted knowing the patient’s age, tumor location, and radiographic characteristics. Treatment is individualized and based on: tumor type, location, and disease stage. Most patients with spinal tumors present with a history of pain often similar in quality and intensity as non-tumor causes of back pain. Spinal neoplasms, especially malignant tumors, require a multidisciplinary approach and are best treated in dedicated cancer centers to mitigate incorrect diagnoses and inappropriate treatment [1]. Accurate diagnosis and prompt treatment are necessary to optimize clinical outcomes.
In this article, we describe the unique features of the most common primary osseous spine tumors in adults regarding: epidemiology, diagnosis, current treatment modalities and potential future therapies. Because primary spinal tumors are uncommon, level I and II data is scarce though novel treatment strategies are emerging. Medical and orthopaedic oncologists and spine surgeons therefore should have a fundamental knowledge of the current state of literature pertaining to this topic.
Materials and Methods
A literature review was conducted using PubMed and EBSCO. Multiple search queries for relevant articles between 2014 to present were included. Preference was given to recent articles with clinical evidence, current treatment, diagnostic modalities and/or future potential therapies and diagnostic strategies.
Benign tumors
Hemangioma: Hemangiomas are hamartomas, normal tissue in an abnormal location, composed of blood vessels, either capillary or cavernous. Neurologic deficits may occur without vertebral collapse, due to expansion and impingement on the neural elements. Vertebral Hemangiomas (VH) are located in the vertebral body and may extend into the pedicles and neural arches (Table 1). Radiographically, the lesion is less radio dense than the surrounding bone conferring a coarsened or “jail bar” trabecular appearance (Figure 1) while on axial CT it is described as “honeycombed” (Figure 2). Bone enlargement and cortical thinning may be observed. Technetium99 pyrophosphate bone scintigraphy (99mTC) usually reveals mildly increased uptake, unless pathologic fracture supervenes in which case uptake is intense. The characteristic MR appearance is mottled and unlike other osseous tumors, is hyperintense on T1 and T2 pulse weighted sequences [2]. They enhance diffusely with gadolinium administration.
![]()
Tumor
Age/Gender
% of Primary Spinal Tumors
Location
Unique imaging features
Hemangioma
4th-6th decade [7]
Female (slight preference) [7]30% of primary spinal tumors7
Observed in 11% of post mortems7No preference
Vertebral bodies“Jail bar” Striations
“Honeycombed” Multiple lesionsGiant Cell Tumor
2nd-4th decades
peak incidence is 3rd decade [12]
Female predilection [12]5% of primary spinal tumors [7,8]
Thoracic and lumbosacral
Vertebral bodies 2nd most common tumor of sacrum
4.9% of GCT’s occur in sacrum [12,71]Aneurysmal features
Heterogeneous
MRI hypointense on T1 and T2 pulse weighted sequences due to hemosiderinChordoma
4th-6th decades
Male (2:1)2-4 % of primary spinal tumors Rare: 0.51-0.8 patients per 100,000141,142
50% sacrum and coccyx [7]
35% skull base
15% mobile spine [7]
Youth: spheno-occipital predilection [39,64]
Vertebral bodies
Most common primary sacral
Tumor [64]Subtle on radiography best seen on lateral radiograph
Midline location Anterior soft tissue extension
Hyperintense on T2 and STIR images and hypointense on T1Multiple Myeloma/Plasmacytoma
5th-6th decade
Male (2:1)20-30 % of primary spinal tumors
Thoracic [7,73]
Vertebral bodiesDiffuse osteopenia
“punched out or moth eaten appearance”
Soft tissue extensionChondrosarcoma
3rd-4th decade
Male (2:1)Most common primary malignant spinal tumor [7,74]
7-12% of primary spinal tumors
10% of CS occur in axial spine [143]Thoracic
Matrix producing (arcs and rings)
Expansile soft tissue extension
Lobular appearance on MROsteosarcoma
3rd-6th decades
Male (slight)
Spinal OS more common in older patients [93,120]<5% of primary spinal tumors
Rare in spine (0.3-3.2 % of all OS) [93,119]
Lumbosacral predilection [120]
Vertebral bodies;
primary involvement of posterior elements seen in 10-17% of cases [144]Matrix producing “cloud like” Destructive with soft tissue extension
Venous involvement common in sacroiliac regionGCT: Giant cell tumor; CS: Chondrosarcoma; OS: Osteosarcoma.
Table 1: Spinal tumor type and its associated demographics.
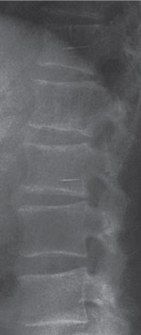
Figure 1: Radiographically VHs may demonstrate a coarsened or “jail bar”
trabecular appearance.
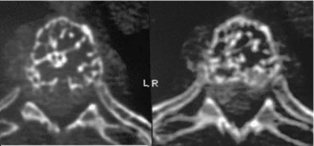
Figure 2: VH. Axial CT demonstrating “honeycombed” appearance of
vertebral body.
Most VH are asymptomatic and treated non-operatively [3]. VH that cause pain, neurologic deficits, or pathologic fracture were traditionally treated by excision following embolization [3,4] (Table 2). In a multicentric study reviewing intralesional resection for symptomatic VH, Goldstein et al. demonstrated tumor recurrence in 2.9% at a mean follow-up of 3.9 years. These results were confounded by treatment variability with 35% receiving preoperative embolization, 10% adjuvant radiation, and 81% undergoing posterior alone surgery [5]. Radiation Therapy (RT) has been advocated for patients with mild pain and minimal bone destruction [4]. When used alone for patients with neurologic symptoms, 82% had complete recovery of their motor and sensory function [6]. In a recent metaanalysis of 197 VH cases, Piper et al. advocated both (RT) and surgery demonstrating a decrease in tumor recurrence [4].
![]()
Table 2: Spinal tumor type and its prospective treatment(s).
Benign aggressive tumors
Giant cell tumor: Giant Cell Tumors (GCT) are benign albeit aggressive lesions composed of bland stromal cells and multinucleated giant cells [7,8]. Patients present with pain and 50% have neurologic symptoms [9]. Radiographically, GCT has a variable appearance. Geographic lysis and expansile growth with cortical thinning may be seen along with a soft tissue mass. Pathologic fracture may occur with extension into adjacent discs and vertebrae [10] (Figure 3 and 4). Pseudotrabeculations may be observed; mineralization is generally absent. In the sacrum, direct extension across the sacroiliac joint into the ilium has been described [11]. Scintigraphically, uptake is heterogeneous, but is generally increased. Aneurysmal changes are relatively common in GCT [12]. MR appearance is variable demonstrating hypointense signal on T1 and T2 pulse-weighted sequences in the solid components due to hemosiderin deposits [12]. Cystic areas are hyperintense on T2 imaging; fluid-fluid levels may be observed [12].
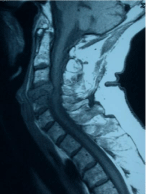
Figure 3: GCT. Sagittal view of the cervical spine, T1 weighted imaging
demonstrating hypointense signal, soft tissue mass, and extension into
adjacent vertebrae.
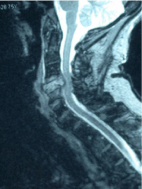
Figure 4: GCT. Sagittal view of the cervical spine, T2 weighted imaging
demonstrating hypointense signal, soft tissue mass, and extension into
adjacent vertebrae.
Biopsy and staging are required as up to 14% of patients with GCT may develop pulmonary metastasis [13,14]. Traditionally, en bloc resection with decompression and stabilization was the preferred treatment resulting in lower recurrence rates and increased survival [8,13,15] (Table 2). Boriani et al stratified surgical resection for GCT of the mobile spine based on Enneking stage. For stage 2 GCTs, intralesional curettage resulted in local recurrence of 6% at 5 years while stage 3 lesions recurred in 61%. Stage 3 lesions treated with en bloc resection recurred in 10% of patients [14]. The treatment of sacral GCTs is complex though traditionally resection with tumor free margins was the goal, with recurrence rates as high as 47% with intralesional resection [16].
Tumor free margins are often not possible due to tumor extent and the risk of permanent neurologic injury. Radiotherapy has been used in select cases where residual macroscopic disease is present or tumor resection is not possible. There is conflicting data on its efficacy and the risk of sarcomatous transformation is reported to be as high as 11% [8,9,11,16-19] Selective Arterial Embolization (SAE) has can reduce intra-operative blood loss, decrease the rate of tumor progression, provide significant pain relief, and alleviate neurologic symptoms [8]. Serial Arterial Embolization (SAE) is used as primary treatment in inoperable lesions. In a review by He et al. SAE of pelvic and sacral GCTs resulted in local disease control and overall survival in 75% and 81.8% of patients respectively [20]. Domovitov et al. compared intralesional resection of sacral GCTs with and without adjuvant XRT and/or SAE. They found significantly fewer local recurrences in patients who underwent neoadjuvant XRT or SAE compared to resection alone [21].
Adjuvant therapy combined with en bloc and subtotal resection may reduce the risk of recurrence. Xu et al. combined surgery with bisphosphonate therapy in treating sacral GCTs observing recurrence in 10.53% of patients in the bisphosphonate group and 47.75% in the control group [22]. In studies involving the mobile spine several authors found increased Recurrence Free Survival (RFS) with adjuvant bisphosphonate therapy and resection [23,24]. Denosumab has been shown to improve neurological symptoms and pain control, and restore bone as single therapy for inoperable GCTs [8,25-27]. Neoadjuvant treatment with denosumab decreases tumor size, blood loss, and defines tumor borders facilitating complete excision [25,26,28]. Lim et al. studied intralesional resection of GCTs and demonstrated 95% recurrence-free survival with adjuvant denosumab vs. 70.3% without [28].
Chordoma
Chordoma arises from notochord remnants. Microscopically, multi-vacuolated physaliferous cells are seen in a bluish myxoid background (Figure 4). Dedifferention is reported and portends a worse prognosis [29]. Patients present with constipation and difficulty sitting though diagnosis is often delayed [30,31]. Metastases occur late in the disease in 10-43 % of patients, principally lungs and liver, although other locations are described [32-35]. Metastatic disease was most common in the youngest patients (P=0.07), and was 2.5 times more frequent for patients with local recurrence (26.3%) compared to those without (10.8%) (P=0.003) [36].
Radiographs are difficult to interpret although lateral radiographs may show a large soft tissue component that is better appreciated on CT or MR. In the mobile spine, chordoma can be expansile and sclerotic, “ivory vertebra” [37]. CT better demonstrates osseous destruction and soft tissue disease extension that often displaces the rectum anteriorly; the fat plane between the mass and the rectum is often preserved although in neglected or recurrent cases, direct rectal involvement may occur (Figures 5 and 6). Inappropriate biopsy through the rectum can also obliterate this important soft tissue plane. Amorphous calcifications have been noted on CT32 along with tumor extension into the spinal canal in 60% of cases [35]. The mass appears uniformly hyperintense on T2 pulse weighted sequences on MR. It has a low to intermediate signal intensity on corresponding T1 images [12].
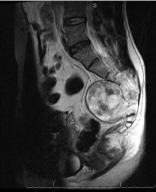
Figure 5: Chordoma. Axial view T2 weighted image with marked
hyperintensity involving and surrounding the sacrum. The fat plane between
the mass and the rectum is preserved.
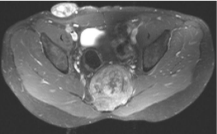
Figure 6: Chordoma. Sagittal T1 weighted image with low to intermediate
signal. The bony architecture of the sacrum has been disrupted however, the
fat plane between the tumor and the rectum is intact.
Treatment is surgical and despite wide margins on gross and microscopic examination, local recurrence is common [38,39]. Intralesional or marginal excisions are associated with high rates of local recurrence [31,33,40] and these patients may have a worse prognosis [39]. Meticulous planning should be undertaken prior to tumor resection and should carefully assess bone and soft tissue extension with particular attention to the level of sacral involvement [38]. Preoperative embolization can reduce surgical blood loss and may be a treatment option in unresectable cases [38,41]. For tumors below the level of S2, a posterior approach is preferred [38]. Tumors above S2 require a combined anterior and posterior approach to mobilize the iliac vessels and omentum and to perform a colostomy [38]. The omentum decreases the dead space along with an acellular dermis or synthetic mesh to prevent rectal prolapse. Gluteal flaps or mobilization of a musculofasciocutaneous rectus abdominis flap may be necessary for wound closure [42,43]. There is a delicate balance between preserving nerve roots and removing adequate tissue to achieve local disease control [39]. Neurological function is highly variable amongst patients treated with sacrectomy [38]. If the S3 nerve roots can be preserved bilaterally, normal bowel and bladder function can be expected although this varies [31,44-46]. Reconstruction following sacrectomy may require spinopelvic fixation, posterior pelvic ring stabilization, and anterior column support due to gross instability [38,47,48].
Adjuvant radiotherapy combined with surgery or alone for palliative care is useful [49,50-52]. Park et al demonstrated that localized radiation after en bloc resection yielded five and ten-year survivorship of 93% and 91%, respectively [38,53]. Radiation seems to confer longer continuous disease-free survival than patients who do not have adjuvant radiotherapy. For those unfit for surgery, radiation therapy can be used as sole therapy [38]. Chen et al demonstrated a 78% five-year survival rate with high dose radiation alone [38,54].
Chordoma is chemotherapy resistant however new biologic pathways are being explored and improved systemic treatments may be available in the future, particularly for advanced disease [55]. There are several new and emerging treatments for patients with chordoma, that are summarized in (Table 2) [38,55-64]. Presently, wide local excision with or without radiation is the preferred treatment while systemic agents are reserved for clinical trials, inoperable or recurrent and metastatic cases.
Multiple Myeloma/Plasmacytoma
Multiple Myeloma (MM) is a clonal proliferation of plasma cells in bone marrow that may be associated with end organ damage. More than 80% of MM patients will develop clinically detectable bone disease; 60% will develop a pathological fracture during the course of the disease [65,66]. Pain, deformity, and neurological deficits are common [67,68].
Radiographs demonstrate well defined lytic “punchedout” lesions usually involving multiple spinal levels (Figure 7). Compression fractures and diffuse osteopenia are due to the upregulation of osteoclast activity through various ligands in the MM microenvironment ie. RANK Ligand (RANKL) [68]. Osteoblast formation is inhibited by MM cell-derived factors, (DKK-1) protein, that inhibits the Wnt pathway resulting in lytic lesions that do not remodel [68,69]. Vertebral lesions may not be apparent radiographically until substantial bone loss has occurred, thus, whole-body low-dose CT is preferred due to its increased sensitivity to detect both osseous and extraosseous lesions.69 MRI is sensitive for detecting intraosseous tumor extent and soft tissue extension [70]. 18F-FDG PET/CT is as sensitive as MRI in identifying solitary plasmacytoma and tumor infiltrated bone marrow [70]. PET/CT is useful for Myeloma (MM) staging and tumor surveillance. Up to 40% of patients initially diagnosed with solitary plasmacytoma have additional lesions diagnosed on PET/CT [70].
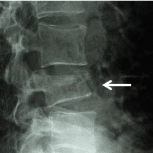
Figure 7: Multiple myeloma. Radiographically MM may demonstrate punched
out lesions or in the spine compression fractures and vertebra planae.
MM is best managed by a multidisciplinary approach involving systemic regimens and possibly bone marrow transplantation that is beyond the scope of this review. Disease extent dictates therapy. For example, patients with solitary plasmacytoma are treated with radiation therapy alone achieving local disease control in 96% of cases [71]. Bisphosphonates are the mainstay of MM bone disease therapy and should be initiated in patients with or without osteolytic lesions and in MM precursor diseases with associated osteoporosis [68]. Intravenous pamidronate and zoledronic acid have been shown to increase overall survival, alleviate bone pain, and decrease Skeletal Related Events (SREs) [68]. Recombinant Osteoprotegerin (OPG) modulates the RANK/RANK-L differentiation of osteoclasts and decreases the number of SREs with efficacy similar to pamidronate (body). Raje et al. demonstrated that denosumab was not inferior to zoledronic acid in decreasing time to the first skeletal related event in newly diagnosed MM patients [72]. Diffuse osteoporosis and pathologic fractures are associated with significant pain, deformity and overall debility. To mitigate these effects, The International Myeloma Working Group recommends early cement augmentation (vertebroplasty) in MM patients with symptomatic vertebral compression fractures.. Prospective treatments for MM can be found in Table 2 [73-91].
Chondrosarcoma
Chondrosarcoma (CS) is a slow-growing, heterogenous group of primary malignant bone tumors characterized by hyaline cartilage formation [92]. Histopathologically, CS can be low, intermediate, or high grade depending on the degree of cellularity, atypia, necrosis and presence of mitoses. There are several disease subcategories including: dedifferentiated, mesenchymal and clear cell variants. CS can arise from benign antecedent tumors such as osteochondromas and enchondromas or, rarely, pagetoid lesions and bone exposed to prior radiation. In general, CS invades locally and the higher the tumor grade, the more likely distant metastases occur, most commonly to the lungs. The most common presenting symptom is pain however neurologic symptoms have been reported in nearly half of patients [93]. Approximately 10% of CS arise in the mobile spine and may be prodigious before causing symptoms (Table 1) [94].
Radiographs show osseous destruction and cartilage matrix reminiscent of arcs and rings, flocculations or stippled calcifications. Bone scintigraphy reveals increased radiotracer uptake. The uptake pattern is more often heterogeneous in malignant cartilage tumors and homogeneous in benign lesions [95]. CT delineates the degree of cortical destruction and matrix production (Figure 8). MR demonstrates a lobular growth pattern that is hyperintense on T2 pulse weighted and STIR images and hypointense on corresponding T1 images. Septal or diffuse enhancement may be observed with gadolinium administration.
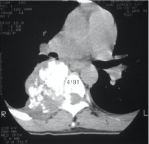
Figure 8: Chondrosarcoma. Axial CT demonstrates cortical destruction and
calcific matrix production.
These tumors are resistant to both radiotherapy and chemotherapy [92,96]. Radiation remains a potential option for incomplete resections, high grade lesions, or palliative care [97]. For this reason, wide local excision alone is the most effective treatment for this disease [94,98-104]. Chen et al performed a retrospective study that stratified and matched primary spinal CS patients by tumor grade and compared outcomes treating CS with surgery alone versus surgery and radiation [105]. Their data demonstrated that patients with low grade tumors treated with surgery alone had better outcomes although those with high grade lesions seemed to benefit from radiation as well [105]. Shamesh et al. demonstrated acceptable results treating low grade chondrosarcoma with intralesional resection [106].
Although systemic therapies have been ineffective for patients with CS, many new potential treatment pathways have emerged. Studies regarding novel targeting microRNA, glucose-metabolism, cyclin-dependent kinase, matrix metalloproteinase, integrins, herbal compounds, adipokines, growth factors and many other molecules are in pre-clinical or early clinical phase trials [92,97]. There are inherent challenges in treating CS with chemotherapy, including the so-called “two-hit” scenario which suggests that more than one pathway is responsible for malignant transformation [97]. Chemotherapy may be used in select cases for example, unresectable tumors that have an estimated five year survival of only 2% [97,107]. A review of new and emerging chemotherapy strategies and pathways are summarized in Table 2 [107-116]. IDH inhibitors have recently been touted as a treatment strategy for CS due to the presence of driver mutations in this pathway and promising results with other tumors bearing similar mutations including leukemia and glioma [107,111,117]. Unfortunately, once malignant progression has occurred in CS, the IDH pathway is no longer thought to be essential for replication [107,111,112]. For this reason IDH targeting therapies may not be as useful in treating CS as once thought [107].
Osteosarcoma
Osteosarcoma is a malignant tumor of bone that produces osteoid [118]. It may be secondary to other conditions such as Paget disease or areas of the spine previously irradiated. Although Paget disease commonly involves the spine, secondary osteosarcoma in this location is very rare [118]. Osteosarcoma of the spine is associated with significant morbidity, early metastases and death [119,120]. Patients with osteosarcomatosis (metachronous or synchronous disease) or patients with relapsed disease may have spinal involvement at multiple levels. As with most spine tumors, pain is the hallmark symptom often accompanied by neurologic deficits [93]. Generally speaking, OS survivorship has plateaued over the past few decades with minimal advances in chemotherapy. Survival is much lower in the axial skeleton [118,121].
Radiographically, tumors are primarily blastic and may appear as an ivory vertebra but may also be predominantly lytic or mixed (blastic and lytic). The tumor begins in the vertebral body and usually has an extraosseous component at the time of diagnosis that is best seen on MR. Dural compression is frequently observed in patients with neurologic deficits. Microscopically, there are many subtypes: osteoblastic (conventional), fibroblastic, chondroblastic, telangiectatic, and small cell. Despite the histologic heterogeneity, the common thread between histologic subtypes is the presence of malignant cells making osteoid. The cells are large and spindled with hyperchromatic nuclei, varying degrees of mitotic activity and cellular and nuclear pleomorphism. Vascular invasion is a prominent feature of pelvic osteosarcoma.
Generally, patients at the extremes of age do poorly. Often older patients, due to comorbidities, cannot tolerate chemotherapy and in those than can, it is difficult to predict tumor response [55]. Patients with axial tumors in general, especially the spine, have a worse prognosis than patients with appendicular lesions. In one study only one patient in twenty-seven was alive at the latest follow-up [93]. The mean survival was 14 months; six patients were paraplegic and three were quadriplegic at the time of death. Dekutoski et al. recently pooled data from twelve spine referral centers studying 58 patients with primary spine osteosarcoma with a mean age of 36 years [122]. An improvement in survival was observed with en bloc resection with a mean survival of 6.8 years compared to those with intralesional resection with a mean survival of 3.7 years [122]. Although 30% of the cohort had local recurrence and 41% were dead at latest follow up, these results were better than those in previous studies [122]. These results were ascribed to improved resection techniques and more aggressive chemotherapy [122]. In their study, age, previous surgery, biopsy method, tumor size, spine level, and chemotherapy timing did not significantly affect recurrence or survival [122]. Previous studies cited local recurrence, secondary osteosarcomas (related to previous radiation or Paget’s disease) and possibly pathologic fractures as negative risk factors for survival [122]. There is a careful balance between preserving function and obtaining an optimal oncologic result [122].
Patients receiving radiation therapy have worse outcomes likely due to patient selection bias, nevertheless, radiation is generally reserved for unresectable tumors and palliative care [55]. Patients with spinal OS are more likely to receive radiation due to the nature of the disease, especially when symptomatic cord compression is present [120]. External beam or proton beam radiation therapy is recommended for patients with gross or residual microscopic disease following resection [55]. The most important survival indicator however is tumor response to neo-adjuvant chemotherapy as measured by tumor necrosis [123,124]. Patients with greater than 90% tumor necrosis after chemotherapy are statistically more likely to survive than those with less tumor necrosis [123,124]. The current chemotherapy OS regimen includes: cisplatin, doxorubicin, and methotrexate [55]. Methotrexate is generally not used in older patients due to its toxicity profile [55,118]. There are several new or emerging treatments and pathways that have garnered interest in osteosarcoma treatment which are summarized in Table 2 [125-140].
Discussion
Primary spine bone tumors in adults are challenging to diagnose and treat for surgeons and oncologists. Resection and preservation of function is the goal and worse outcomes compared to patients with appendicular tumors are expected. Clinical, radiographic and pathologic correlation in a multidisciplinary setting is critical to properly diagnose and treat these conditions. Appropriate workup and treatment is optimized in tertiary spine and tumor referral centers. Surgery, chemotherapy, evolving immunologic and targeted therapies as well as stereotactic EBRT and proton therapy are utilized to optimize care. Still current therapeutic strategies result in significant morbidity and mortality and local disease recurrence and systemic relapse are common despite chemotherapy and advanced surgical techniques.
References
- Mankin CJ, Simon MA. The hazards of the biopsy, revisited. Members of the Musculoskeletal Tumor Society. J Bone Joint Surg Am. 1996; 78: 656-663.
- Ross JS, Masaryk TJ, Modic MT, et al. Vertebral hemangiomas: MR imaging. Radiology. 1987; 165: 165-169.
- Acosta FL, Sanai N, Chi JH, et al. Comprehensive Management of Symptomatic and Aggressive Vertebral Hemangiomas. Neurosurgery Clinics of North America. 2008; 19: 17-29.
- Piper K, Zou L, Li D, et al. Surgical Management and Adjuvant Therapy for Patients With Neurological Deficits From Vertebral Hemangiomas: A Meta- Analysis. Spine (Phila Pa 1976). 2020; 45: E99-E110.
- Goldstein CL, Varga PP, Gokaslan ZL, et al. Spinal hemangiomas: results of surgical management for local recurrence and mortality in a multicenter study. Spine (Phila Pa 1976). 2015; 40: 656-664.
- Asthana AK, Tandon SC, Pant GC, et al. Radiation therapy for symptomatic vertebral haemangioma. Clin. Oncol. (R Coll Radiol). 1990; 2: 159-162.
- Chi JH, Bydon A, Hsieh P, et al. Epidemiology and Demographics for Primary Vertebral Tumors. Neurosurgery Clinics of North America. 2008; 19: 1-4.
- Luksanapruksa P, Buchowski JM, Singhatanadgige W, et al. Management of spinal giant cell tumors. The Spine Journal. 2016; 16: 259-269.
- Sanjay BKS, Sim FH, Unni KK, et al. Giant-cell tumors of the spine. J. Bone and Joint Surg. 1993; 75B: 148-154,
- Si M-J, Wang C-G, Wang C-S, et al. Giant cell tumours of the mobile spine: characteristic imaging features and differential diagnosis. La Radiologia Medica. 2014; 119: 681-693.
- Smith TP, Gray L, Weinstein JN, et al. Preoperative transarterial embolization of spinal column neoplasms. J Vasc Interv Radiol. 1995; 6: 863-869.
- Murphey MD, Nomikos GC, Flemming DJ, et al. Imaging of giant cell tumor and giant cell reparative granuloma of bone: radiologic-pathologic correlation. Radiographics. 2001; 21: 1283-1309.
- Donthineni R, Boriani L, Ofluoglu O, et al. Metastatic behaviour of giant cell tumour of the spine. Int Orthop. 2009; 33: 497-501.
- Boriani S, Bandiera S, Casadei R, et al. Giant Cell Tumor of the Mobile Spine. Spine. 2012; 37: E37–E45.
- Charest-Morin R, Fisher CG, Varga PP, et al. En Bloc Resection Versus Intralesional Surgery in the Treatment of Giant Cell Tumor of the Spine. SPINE, 2017; 42: 1383-1390.
- Leggon RE, Zlotecki R, Reith J, et al. Giant Cell Tumor of the Pelvis and Sacrum. Clinical Orthopaedics and Related Research. 2004; 423: 196-207.
- Cambell CJ, Bonfiglio M. Aggressiveness and malignant in giant cell tumors of bone. In: Price CHG, Ross FGM, eds. Bone: certain aspects of neoplasia. London: Butterworth and Colston Research Society. 1973: 15-40.
- Harwood AR, Fornasier VL, Rider WD. Supervoltage irradiation in the management of giant cell tumors of bone. Radiology. 1977; 125: 223-226.
- McGrath PJ. Giant cell tumor of bone: an analysis of 52 cases. J. Bone and Joint Surg. 1972; 54B: 216-229,
- He SH, Xu W, Sun ZW, et al. Selective Arterial Embolization for the Treatment of Sacral and Pelvic Giant Cell Tumor: A Systematic Review. Orthop Surg. 2017; 9: 139-144.
- Domovitov SV, Chandhanayingyong C, Boland PJ, et al. Conservative surgery in the treatment of giant cell tumor of the sacrum: 35 years’ experience. J Neurosurg Spine. 2016; 24: 228-240.
- Xu W, Wang Yu, Wang J, et al. Long-term administration of bisphosphonate to reduce local recurrence of sacral giant cell tumor after nerve-sparing surgery, Journal of Neurosurgery: Spine. 2017; 26: 716-721.
- Xu W, Li X, Huang W, et al. Factors Affecting Prognosis of Patients with Giant Cell Tumors of the Mobile Spine: Retrospective Analysis of 102 Patients in a Single Center. Annals of Surgical Oncology. 2012; 20: 804–810.
- Pannu CD, Kandhwal P, Raghavan V, et al. Role of Bisphosphonates as Adjuvants of Surgery in Giant Cell Tumor of Spine. Int J Spine Surg. 2018; 12: 695-702.
- Thornley P, Habib A, Bozzo A, et al. The Role of Denosumab in the Modern Treatment of Giant Cell Tumor of Bone. JBJS Reviews. 2017; 5: e4.
- Boriani S, Cecchinato R, Cuzzocrea F, et al. Denosumab in the treatment of giant cell tumor of the spine. Preliminary report, review of the literature and protocol proposal. Eur Spine J. 2020; 29: 257-271.
- Goldschlager T, Dea N, Boyd M, et al. Giant cell tumors of the spine: has denosumab changed the treatment paradigm?. J Neurosurg Spine. 2015; 22: 526-533.
- Lim CY, Liu X, He F, et al. Retrospective cohort study of 68 sacral giant cell tumours treated with nerve-sparing surgery and evaluation on therapeutic benefits of denosumab therapy. Bone Joint J. 2020; 102-B: 177-185.
- Catton C, O’Sullivan B, Bell R, et.al. Chordoma: Long-term follow-up after radical photon irradiation. Radiother Oncol. 1996; 41: 67-72.
- Bethke KP, Neifeld JP, Lawrence W. Diagnosis and management of sacrococcygeal chordoma. Journal of Surgical Oncology. 1991; 48: 232-238.
- Cheng EY, Ozerdemoglu RA, Transfeldt EE, et al. Lumbosacral Chordoma: Prognostic factors and treatment. Spine. 1999; 24: 1639-1645.
- Firoozniz H, Golimbu C, Rafii M, et al. Computed tomography of spinal chordomas. J. Compute Assist Tomogr. 1986; 10: 45-50.
- Jeanrot C, Vihn TS, Anract P, et al. Sarcal Chordoma: retrospective review of 11 surgically treated cases. Revue de Chirurgie Orthopedique et Reparatrice de I Appareil Moteur. 2000; 86: 684-693.
- Bjornsson J, Wold LE, Ebersold MJ, et al. Chordoma of the mobile spine: a clinicopathologic analysis of 40 patients. Cancer. 1993; 71: 735-740.
- Meyer JE, Lepke RA, Lindfors KK, et al. Chordomas: Their CT appearance in the cervical, thoracic and lumbar spine. Radiology. 1984; 153: 693-696.
- Young VA, Curtis KM, Temple HT, et al. Characteristics and Patterns of Metastatic Disease from Chordoma. Sarcoma. 2015; 2015: 517657.
- Braun RA, Milito CFRB, Goldman SM, et al. Ivory vertebra: imaging findings in different diagnoses. Radiol Bras. 2016; 49: 117-121.
- Pillai S, Govender S. Sacral chordoma: A review of literature. J Orthop. 2018; 15: 679-684.
- Denaro L, Berton A, Ciuffreda M, et al. Surgical management of chordoma: A systematic review. J Spinal Cord Med. 2018; 1-16.
- Ozaka T, Toriyama S. Treatment and prognosis of sacrococcygeal chordoma: study of bone tumor registry in Japan [In Japanese]. Nippon Seikeigeka Gakkai Zzasshi. 1989; 63: 240-244.
- Mavrogenis AF, Rossi G, Rimondi E, et al. Embolization of bone tumors. Orthopedics. 2011; 34: 303-310.
- Garofalo F, di Summa PG, Christoforidis D, et al. Multidisciplinary approach of lumbo-sacral chordoma: From oncological treatment to reconstructive surgery. J Surg Oncol. 2015; 112: 544-554.
- Dasenbrock HH, Clarke MJ, Bydon A, et al. Reconstruction of extensive defects from posterior en bloc resection of sacral tumors with human acellular dermal matrix and gluteus maximus myocutaneous flaps. Neurosurgery. 2011; 69: 1240-1247.
- Guttenberg B, Norlen L, Stener B, et al. Neurologic evaluation after resection of the sacrum. Invest Urol. 1975; 13: 183-188.
- Samson IR, Springfield DS, Suit HD, et al. Operative treatment of sacrococcygeal chordoma. A review of twenty-one cases. J Bone Joint Surg. 1993; 75A: 1476-1484.
- Ahrens S, Hoffman C, Jabar S, et al. Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: The CESS 86 experience. Cooperative Ewing Sarcoma Study. Medical and Pediatric Oncology. 1999; 32: 186-195.
- Fourney DR, Gokaslan ZL. Spinal instability and deformity due to neoplastic conditions. Neurosurg Focus. 2003; 14: e8.
- Ishida W. Spinopelvic reconstruction following lumbosacral tumor resection. Department of Neurosurgery, Johns Hopkins University, School of Medicine WScJ. 2016; 1: 25-30.
- Azzarelli A, Quagliuolo V, Cerasoli S, et al. Chordoma: natural history and treatment results in 33 cases. J Surg Oncol. 1988; 37: 185-191.
- Breteau N, Demasure M, Favre A, et al. Fast neutron therapy for inoperable or recurrent sacrococcygeal chordomas. Bull Cancer Radiother. 1996; 83: 1425-1455.
- Chandawarkar RY. Sacrococcygeal chordoma: Review of 50 consecutive patients. World J Surg. 1996; 20: 717-719.
- Coley BL, Higinbothan NL, Bowden L. Endothelioma of bone (Ewing’s sarcoma). Ann Surg. 1948; 128: 533-560.
- Park L, Delaney TF, Liebsch NJ, et al. Sacral chordomas: Impact of highdose proton/photon-beam radiation therapy combined with or without surgery for primary versus recurrent tumor. Int J Radiat Oncol Biol Phys. 2006; 65: 1514-1521.
- Chen YL, Liebsch N, Kobayashi W, et al. Definitive high-dose photon/proton radiotherapy for unresected mobile spine and sacral chordomas. Spine (Phila Pa 1976). 2013; 38: E930-E936.
- Whelan JS, Davis LE. Osteosarcoma, Chondrosarcoma, and Chordoma. J Clin Oncol. 2018; 36: 188-193.
- Meng T, Jin J, Jiang C, et al. Molecular Targeted Therapy in the Treatment of Chordoma: A Systematic Review. Front Oncol. 2019; 9: 30.
- Heery CR. Chordoma: The Quest for Better Treatment Options. Oncol Ther. 2016; 4: 35-51.
- Patel SS, Schwab JH. Immunotherapy as a Potential Treatment for Chordoma: a Review. Curr Oncol Rep. 2016; 18: 55.
- Magnaghi P, Salom B, Cozzi L, et al. Is a New Therapeutic Approach in Chordoma with a Unique Ability to Target EGFR and Brachyury. Mol Cancer Ther. 2018; 17: 603-613.
- Migliorini D, Mach N, Aguiar D, et al. First report of clinical responses to immunotherapy in 3 relapsing cases of chordoma after failure of standard therapies. Oncoimmunology. 2017; 6: e1338235.
- Frezza AM, Botta L, Trama A, et al. Chordoma: update on disease, epidemiology, biology and medical therapies. Curr Opin Oncol. 2019; 31: 114-120.
- Stacchiotti S, Sommer J. Chordoma Global Consensus Group. Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol. 2015; 16: e71-83.
- Liu T, Shen JK, Choy E, Zhang Y, Mankin HJ, Hornicek FJ, et al. CDK4 expression in chordoma: A potential therapeutic target. J Orthop Res. 2018; 36: 1581-1589.
- Mirra J NS, Della Rocca C, et al. Chordoma. In: Fletcher CD UK, Mertens F, (ed) Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press. 2002: 316-317.
- Roodman GD. Pathogenesis of myeloma bone disease. J Cell Biochem. 2010; 109: 283-291.
- Melton LJ, 3rd, Kyle RA, Achenbach SJ, et al. Fracture risk with multiple myeloma: a population-based study. J Bone Miner Res. 2005; 20: 487-493.
- Delauche-Cavallier MC, Laredo JD, Wybier M, et al. Solitary plasmacytoma of the spine. Long-term clinical course. Cancer. 1988; 61: 1707-1714.
- Heusschen R, Muller J, Duray E, et al. Molecular mechanisms, current management and next generation therapy in myeloma bone disease. Leuk Lymphoma. 2018; 59: 14-28.
- Walker RC, Brown TL, Jones-Jackson LB, et al. Imaging of multiple myeloma and related plasma cell dyscrasias. J Nucl Med. 2012; 53: 1091-1101.
- Derlin T, Bannas P. Imaging of multiple myeloma: Current concepts. World J Orthop. 2014; 5: 272-282.
- Hsu W, Kosztowski TA, Zaidi HA, et al. Multidisciplinary management of primary tumors of the vertebral column. Curr Treat Options Oncol. 2009; 10: 107-125.
- Raje N, Terpos E, Willenbacher W, et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: an international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018; 19: 370-381.
- Ropper AE, Cahill KS, Hanna JW, et al. Primary vertebral tumors: a review of epidemiologic, histological and imaging findings, part II: locally aggressive and malignant tumors. Neurosurgery. 2012; 70: 211-219.
- Kelley SP, Ashford RU, Rao AS, et al. Primary bone tumours of the spine: a 42-year survey from the Leeds Regional Bone Tumour Registry. European Spine Journal. 2006; 16: 405-409.
- Zangari M, Suva LJ. The effects of proteasome inhibitors on bone remodeling in multiple myeloma. Bone. 2016; 86: 131-138.
- Pennisi A, Li X, Ling W, et al. The proteasome inhibitor, bortezomib suppresses primary myeloma and stimulates bone formation in myelomatous and nonmyelomatous bones in vivo. Am J Hematol. 2009; 84: 6-14.
- Deleu S, Lemaire M, Arts J, et al. Bortezomib alone or in combination with the histone deacetylase inhibitor JNJ-26481585: effect on myeloma bone disease in the 5T2MM murine model of myeloma. Cancer Res. 2009; 69: 5307-5311.
- Hurchla M, Garcia-Gomez A, Hornick MC, et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia. 2013; 27: 430-440.
- Garcia-Gomez A, Quwaider D, Canavese M, et al. Preclinical activity of the oral proteasome inhibitor MLN9708 in myeloma bone disease. Clin Cancer Res. 2014; 20: 1542-1554.
- Oyajobi BO, Garrett IR, Gupta A, et al. Stimulation of new bone formation by the proteasome inhibitor, bortezomib: implications for myeloma bone disease. Br J Haematol. 2007; 139: 434-438.
- Hongming H, Jian H. Bortezomib inhibits maturation and function of osteoclasts from PBMCs of patients with multiple myeloma by downregulating TRAF6. Leuk Res. 2009; 33: 115-122.
- Zangari M, Terpos E, Zhan F, et al. Impact of bortezomib on bone health in myeloma: a review of current evidence. Cancer Treat Rev. 2012; 38: 968- 980.
- Munemasa S, Sakai A, Kuroda Y, et al. Osteoprogenitor differentiation is not affected immunomodulatory thalidomide analogs but is promoted by low bortezomib concentration, while both agents suppress osteoclast differentiation. Int J Oncol. 2008; 33: 129-136.
- Anderson G, Gries M, Kurihara N, et al. Thalidomide derivative CC-4047 inhibits osteoclast formation by down-regulation of PU.1. Blood. 2006; 107: 3098-3105.
- Chang X, Zhu Y, Shi C, et al. Mechanism of immunomodulatory drugs’ action in the treatment of multiple myeloma. Acta Biochim Biophys Sin (Shanghai). 2014; 46: 240-253.
- Scullen T, Santo L, Vallet S, et al. Lenalidomide in combination with an activin A-neutralizing antibody:preclinical rationale for a novel anti-myeloma strategy. Leukemia. 2013; 27: 1715-1721.
- Bolomsky A, Schreder M, Meissner T, et al. Immunomodulatory drugs thalidomide and lenalidomide affect osteoblast differentiation of human bone marrow stromal cells in vitro. Exp Hematol. 2014; 42: 516-525.
- Fulciniti M, Tassone P, Hideshima T, et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood. 2009; 114: 371-379.
- Eda H, Santo L, Wein MN, et al. Regulation of sclerostin expression in multiple myeloma by Dkk-1: a potential therapeutic strategy for myeloma bone disease. J Bone Miner Res. 2016; 31: 1225-1234.
- Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016; 375: 1532- 1543.
- Abdulkadyrov KM, Salogub GN, Khuazheva NK, et al. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br J Haematol. 2014; 165: 814-823.
- Chow WA. Chondrosarcoma: biology, genetics, and epigenetics. F1000 Res. 2018; 7: F1000.
- Shives TC, McLeod RA, Unni K, et al. Chondrosarcoma of the Spine. Journal Bone and Joint Surgery. 1989; 71A: 1158-1165.
- Blaylock RL, Kempe LG. Chondrosarcoma of the cervical spine. Case report. Journal of Neurosurgery. 1976; 44: 500-503.
- Murphey MD, Flemming DJ, Boyea SR, et al. Enchondroma versus Chondrosarcoma in the Appendicular Skeleton: Differentiating Features. Radiographics. 1998; 18: 1213-1237.
- Vergel De Dios AM, Bond JM, Shives TC, et al. Aneurysmal bone cyst: a clinicopathologic study of 238 cases. Cancer. 1992; 69: 2921-2931.
- MacDonald IJ, Lin CY, Kuo SJ, et al. An update on current and future treatment options for chondrosarcoma. Expert Rev Anticancer Ther. 2019; 19: 773-786.
- Camins MB, Duncan AW, Smith J, Marcove RC. Chondrosarcoma of the spine. Spine. 1978; 3: 202-209.
- Formica C, Gavino D, Giudice V. Tumors of the spine. Indications for surgical treatment. Chirurgia Degli Organi di Movimento. 1988; 83: 23-33.
- Fuentes JM, Benezech J. Stratefy of the surgical treatment of primary tumors of the spine. Neuro-Chirurgie. 1989; 35: 323-327.
- Kala M, Chrobok, J, Dvorak P. Surprising finding by surgery for lumbar intervertebral disk herniation. Ewing’s sarcoma. Acta Unic Palacki Olomuc Fac Med. 1987;117: 185-189.
- Kornberg M. Primary Ewing’s sarcoma of the spine. A review and case report. Spine. 1986; 11: 54-57.
- Winderen M, Stenwig AE, Solheim OP, et al. Dynamic bone scintigraphy for evaluation of tumor response after preoperative chemotherapy. A retrospective study of osteosarcoma and Ewing’s sarcoma patients. Acta Orthopaedica Scandinavica. Supplementum. 1999; 285: 11-17.
- Zibis AH, Markonis A, Karantanas AH. Unusual causes of spinal foraminal widening. Review. Review, Tutorial] European Radiology. 2000; 10: 144- 148.
- Chen D, Chen C-H, Zhang L-L, et al. Chondrosarcoma of the Osseous Spine Treated by Surgery With or Without Radiotherapy.ClinicalSpineSurgery. 2018; 31: E310-E316.
- Shemesh SS, Pretell-Mazzini J, Quartin PAJ et al. Surgical treatment of low-grade chondrosarcoma involving the appendicular skeleton: long-term functional and oncological outcomes. Arch Orthop Trauma Surg. 2019; 139: 1659-1666.
- van Maldegem A, Conley AP, Rutkowski P, et al. Outcome of First-Line Systemic Treatment for Unresectable Conventional, Dedifferentiated, Mesenchymal, and Clear Cell Chondrosarcoma. Oncologist. 2019; 24: 110- 116.
- Meijer D, Gelderblom H, Karperien M et al. Expression of aromatase and estrogen receptor alpha in chondrosarcoma, but no beneficial effect of inhibiting estrogen signaling both in vitro and in vivo. Clin Sarcoma Res 2011; 1: 5.
- Cleton-Jansen AM, van Beerendonk HM, Baelde HJ et al. Estrogen signaling is active in cartilaginous tumors: Implications for antiestrogen therapy as treatment option of metastasized or irresectable chondrosarcoma. Clin Cancer Res. 2005; 11: 8028-8035.110. Grifone TJ, Haupt HM, Podolski V, et al. Immunohistochemical expression of estrogen receptors in chondrosarcomas and enchondromas. Int J Surg Pathol. 2008; 16: 31-37.
- Suijker J, Oosting J, Koornneef A, et al. Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget. 2015; 6: 12505-12519.
- Li L, Paz AC, Wilky BA, et al. Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS One. 2015; 10: e0133813.
- Campbell VT, Nadesan P, Ali SA, et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Mol Cancer Ther. 2014; 13: 1259-1269.
- Tiet TD, Hopyan S, Nadesan P, et al. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am J Pathol. 2006; 168: 321-330.
- Schrage YM, Briaire-de Bruijn IH, de Miranda NF, et al. Kinome profiling of chondrosarcoma reveals SRC-pathway activity and dasatinib as option for treatment. Cancer Res. 2009; 69: 6216-6222.
- van Oosterwijk JG, van Ruler MA, Briaire-de Bruijn IH, et al. Src kinases in chondrosarcoma chemoresistance and migration: Dasatinib sensitises to doxorubicin in TP53 mutant cells. Br J Cancer. 2013; 109: 1214-1222.
- Jin Y, Elalaf H, Watanabe M, et al. Mutant IDH1 dysregulates the differentiation of mesenchymal stem cells in association with gene-specific histone modifications to cartilage and bone-related genes. PLoS One. 2015; 10: e0131998.
- Anderson ME. Update on Survival in Osteosarcoma. Orthop Clin North Am. 2016; 47: 283-292.
- Barwick, KW, Huvos AG, Smith J. Primary osteogenic sarcoma of the vertebral column: a clinicopathologic correlation of ten patients. Cancer. 1980; 46: 595-604.
- Arshi A, Sharim J, Park DY, et al. Prognostic determinants and treatment outcomes analysis of osteosarcoma and Ewing sarcoma of the spine. Spine J. 2017; 17: 645-655.
- Zhao J, Dean DC, Hornicek FJ, et al. Emerging next-generation sequencingbased discoveries for targeted osteosarcoma therapy. Cancer Lett. 2020; 474: 158-167.
- Dekutoski MB, Clarke MJ, Rose P, et al. Osteosarcoma of the spine: prognostic variables for local recurrence and overall survival, a multicenter ambispective study. J Neurosurg Spine. 2016; 25: 59-68.
- Ferrari S, Bacci G, Picci P, et al. Long-term follow-up and post-relapse survival in patients with non-metastatic osteosarcoma of the extremity treated with neoadjuvant chemotherapy. Annals of Oncology. 1997; 8: 765- 771.
- Provisor AJ, Ettinger LJ, Nachman LB, et al. Treatment of nonmetastatic osteosarcoma of the extremity with preoperative and postoperative chemotherapy: a report from the Children’s Cancer Group. Journal of Clinical Oncology. 1977; 15: 76-84.
- Subbiah V, Anderson P, Rohren E. Alpha Emitter Radium 223 in High-Risk Osteosarcoma: First Clinical Evidence of Response and Blood-Brain Barrier Penetration. JAMA Oncol. 2015; 1: 253-255.
- Meyers PA, Schwartz CL, Krailo MD, et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival–A report from the Children’s Oncology Group. J Clin Oncol. 2008; 26: 633-638.
- Buddingh EP, Kuijjer ML, Duim RA, et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin Cancer Res. 2011; 17: 2110-2119.
- Koirala P, Roth ME, Gill J, et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci Rep. 2016; 6: 30093.
- Burgess M, Gorantla V, Weiss K, et al. Immunotherapy in sarcoma: future horizons. Curr Oncol Rep. 2015; 17: 1-0.
- Grignani G, Palmerini E, Ferraresi V, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015; 16: 98-107.
- Xie L, Xu J, Sun X, et al. Apatinib for Advanced Osteosarcoma after Failure of Standard Multimodal Therapy: An Open Label Phase II Clinical Trial. Oncologist. 2019; 24: e542-e550.
- Davis, Lara E, et al. Randomized double-blind phase II study of regorafenib in patients with metastatic osteosarcoma.”Journal of Clinical Oncology. 2019; 37: 1424-1431.
- Sayles LC, Breese MR, Koehne AL, et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019; 9: 46-63.
- Chen Y, Wu JJ, Huang L. Nanoparticles targeted with NGR motif deliver c-myc siRNA and doxorubicin for anticancer therapy, Mol. Ther. 2010; 18: 828-834.
- Zhang Y, Peng L, Mumper RJ, et al. Combinational delivery of c-myc siRNA and nucleoside analogs in a single, synthetic nanocarrier for targeted cancer therapy, Biomaterials. 2013; 34: 8459-8468.
- Zhou YB, Shen JK, Yu ZJ, et al. Expression and therapeutic implications of Cyclin-Dependent Kinase 4 (CDK4) in osteosarcoma, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2018; 1864: 1573-1582.
- Grignani G, Palmerini E, Dileo P, et al. A phase II trial of sorafenib in relapsed and unresectable high-grade OS after failure of standard multimodal therapy: an Italian Sarcoma Group study. Ann Oncol. 2012; 23: 508-516.
- Chawla SP, Staddon AP, Baker LH, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas, J. Clin. Oncol. 2012; 30: 78-84.
- Pappo AS, Vassal G, Crowley JJ, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research through Collaboration study, Cancer. 2014; 120: 2448–2456.
- Anderson PM, Bielack SS, Gorlick RG, et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma, Pediatr. Blood Canc. 2016; 63: 1761–1770.
- Higinbothan NL, Phillips RF, Farr HW, et al. Chordoma: thirty-five-year study at Memorial Hospital. Cancer. 1967; 20: 1841-1850.
- McMaster ML, Goldstein AM, Bromley CM, et al. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes and Control. 2001; 12: 1-11.
- Boriani S, DeIure F, Bandiera S, et al. Chondrosarcoma of the mobile spine: report on 22 cases. Spine. 2000; 25: 804-812.
- Wuisman PI, Jutte PC, Ozaki T. Secondary chondrosarcoma in osteochondromas. Medullary extension in 15 of 45 cases. Acta Orthop Scand. 1977; 68: 396-400.
- Schuetze SM, Bolejack V, Choy E, et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cance.r 2017; 123: 90–97.