
Review Article
Austin J Vasc Med. 2014;1(1): 8.
Protein Kinase C and Rho-Associated Coiled-Coil Kinase in Mechanisms of Ca2+ Sensitization in Diabetes-Induced Vascular Smooth Muscle Hypercontractility
Kizub IV1,2*
1Department of Experimental Therapeutics, Institute of Pharmacology and Toxicology of National Academy of Medical Sciences of Ukraine, Ukraine
2Department of Pharmacology, New York Medical College, USA
*Corresponding author: Kizub IV, Department of Experimental Therapeutics, Institute of Pharmacology and Toxicology of National Academy of Medical Sciences of Ukraine, 14 Eugene Pottier St, Kiev, 03680, Ukraine
Received: August 15, 2014; Accepted: September 19, 2014; Published: September 22, 2014
Abstract
Diabetes is a complex syndrome which leads to multiple dysfunctions including vascular disorders. Protein kinase C (PKC) and Rho-associated coiled-coil kinase (ROCK) are important regulatory enzymes mediating signal transduction in a number of vascular functions, including vascular smooth muscle contractility. Many studies have shown that vascular smooth muscle contractile responses are dramatically enhanced in diabetes where in, PKC and ROCK significantly contribute to the process by mediating Ca2+ sensitization of contractile proteins. Both PKC- and ROCK-mediated pathways converge to phosphorylate the inhibitory protein CPI-17 which binds to myosin light chain phosphatase (MLCP) and inhibits its activity. Besides, ROCK phosphorylates myosin light chain phosphatase targeting subunit 1 (MYPT1) which also inhibits the phosphatase activity. Inhibition of MLCP results in a higher level of myosin light chain phosphorylation for any given intracellular level of Ca2+ and activity of the myosin light chain kinase (MLCK). Ca2+ sensitization of smooth muscle, thus, could potentially maintain vascular contraction in diabetes. A link between the ROCK and PKC pathways in diabetic vascular smooth muscle cells has been shown, assuming that the ROCK and RhoA protein are downstream effectors of PKC. Furthermore, collected data suggest that PKC and ROCK are potential therapeutic targets for treating diabetes-related complications in vascular smooth muscle cells.
Keywords: Diabetes; Protein kinase C; Rho kinase; Ca2+ sensitization; Vascular smooth muscle; Vascular tone
Abbreviations
AA: Arachidonic Acid; CaM: Calmodulin; CPI-17: Protein Kinase C-dependent Protein Phosphatase-1 Inhibitor of 17 kDa; DAG: Diacylglycerol; DHAP: Dihydroxyacetone Phosphate; DM: Diabetes Mellitus; EGFR: Epidermal Growth Factor Receptor; ErbB2: Epidermal Growth Factor Receptor B Family Member 2; ERK1/2: Extracellular Signal-regulated Kinases1 and 2; EP: Prostaglandin E Receptor; 5-HT: 5-Hydroxytryptamine; GAP: Glyceraldehyde- 3-phosphate; Gαq/11: Guanosine-5’-triphosphate-binding Proteins Gαq Family Subunit; Gα12/13: Guanosine-5’-triphosphate-binding Proteins Gα12/13 Family Subunits; GDP: Guanosine Diphosphate; GPCR: G-protein Coupled Receptor; Grb-2: Growth Factor Receptor-bound Protein 2; GT: Guanosine-5’-triphosphate; IP3: Inositol-1,4,5-trisphosphate; IP3R: Inositol-1,4,5-trisphosphate Receptor; iPLA2β: Calcium-independent Phospholipase A2β; 12/15-LOX: 12/15-lipoxygenases; LPI: Lysophosphatidylinositol; MEK1/2: Mitogen-activated Protein Kinase Kinases 1 and 2; MLCK: Myosin Light Chain Kinase; MLCP: Myosin Light Chain Phosphatase; mRNA: Messenger Ribonucleic Acid; MYPT1: Myosin-bound Myosin Light Chain Phosphatase Targeting Subunit 1; PA: Phosphatidic Acid; PIP2: Phosphatidylinositol-4,5-bisphosphate; PKC: Protein Kinase C; PLCγ: Phospholipase Cγ; PP1: Protein Kinase C-dependent Protein Phosphatase-1; PP1cδ: Protein Kinase C-dependent Protein Phosphatase-1 Catalytic Subunit; Raf1: Rapidly Accelerated Fibrosarcoma Proto-oncogene protein kinase; Ras: Rat Sarcoma Protein; Rho: Ras Homolog Protein; RhoGAP: Rho GTPase Activating Protein; RhoGDI: Rho Guanine Nucleotide Dissociation Inhibitor; RhoGEF: Rho Guanine-nucleotide Exchange Factors; ROCK: Rho-associated Coiled-coil Kinase; ROS: Reactive Oxygen Species; SMCs: Smooth Muscle Cells; Sch: Src Homology 2 Domain Containing Transforming Protein; SOS: Son of Sevenless Homolog Protein; SR: Sarcoplasmic Reticulum; STZ: Streptozotocin
Introduction
Diabetes mellitus (DM) is a wide-spread syndrome that is rapidly rising in frequency throughout the world. Hyperglycemia and alterations of metabolism are the most severe components of DM [1,2]. Near5-10% of patients with DM have autoimmune type 1 insulin-dependent diabetes, whereas 90-95% have type 2 DM (insulin-independent), which is a consequence of lifestyle patterns contributing to obesity [1,2]. Type 2 DM typically occurs in the context of a cluster of cardiovascular risk factors [1]. DM is known to cause multiple dysfunctions including cardiovascular diseases, which are major causes of mortality, end-stage renal disease, and blindness [3]. The macrovascular manifestations of DM, such as angiopathy, atherosclerosis, medial calcification, and arterial hypertension, have been shown to mostly locate in coronary and carotid arteries [4], cerebral vessels [5], and large peripheral arteries of the lower extremities [5]. The microvascular complications of DM include retinopathy [6], nephropathy [7], and peripheral neuropathy [8]. Increased blood flow and elevated vascular tone have also been documented for DM [3]. A growing body of evidence indicates that endothelial and smooth muscle dysfunctions are present in various regions of the vasculature in both diabetic patients and animal models of DM [9-14].
Hyperglycemia is considered to be a key factor responsible for the development of vascular complications in DM [3,15]. Several hyperglycemia-associated mechanisms, including oxidative stress have been identified to contribute to the development of DM-associated vascular dysfunctions [16-18]. Dysfunctions in the regulation of vascular cells permeability, growth, angiogenesis, and vascular smooth muscle contractility in DM have been shown to involve protein kinase C (PKC) and Rho-associated coiled-coil kinase (ROCK)upregulation [13,18-20].
PKC
PKC is a regulatory enzyme that plays a significant role in signal transduction of several vascular functions [21]. PKC is presented with a family of serine/threonine kinases, with at least known 10 isoforms [21]. Based on homology and sensitivity to the activators, PKC isozymes are classified into three subfamilies: conventional (or classical), novel, and atypical PKC [21]. Classical isozymes include PKC-α, PKC-β1, PKC-β2 and PKC-γ [22,23]. The novel PKC subfamily consists of PKC-δ, PKC-ε, PKC-η, and PKC-θ [24,25]. The atypical isozymes are represented with PKC-ζ and PKC-ι/ [21,22]. In resting cells, PKCs primarily locate in the cytosolic fraction and the enzymes’ translocation to the plasma membrane conventionally has been considered as the hallmark of PKC activation [21]. Interestingly, the former PKC-μ and PKC-ν isoforms are now classified as members of the DAG receptor protein kinase D family [26]. Expression of numerous PKC isozymes (α, β1, β2, γ, ε, η, ζ, δ, and ι/λ) in vascular tissues depends on the animal species, as well as the type and age of the vessel [18,22]. The PKC-α, PKC-β1, PKC-β2, PKC-γ, PKC-δ, PKC-ε, and PKC-ζ isoforms have been shown to be activated or overexpressed in vascular smooth muscle cells (SMCs) and the endothelium of different vascular regions in diabetes [14,27-31].
Hyperglycemia in DM results in an overproduction of reactive oxygen species (ROS) and the ensuing oxidative stress which contributes to the activation of PKC [20,32,33]. Furthermore, there is strong evidence that PKC activation is mediated, at least in part, by induction of oxidative stress and increased production of ROS [27,34,35]. Important to note, PKC is well known to be highly sensitive to oxidative stress [21]. In vascular tissues, PKC activation can also be mediated by diacylglycerol (DAG) [5,20,36] which has been shown to be elevated in DM [27,30,37,38]. Hyperglycemia can enhance the amount of DAG primarily by increasing de novo DAG synthesis from the glycolytic intermediate dihydroxyacetone phosphate (DHAP), as well as by inducing phosphorylation of the phospholipase C (PLCγ) [20, 38] (Figure).
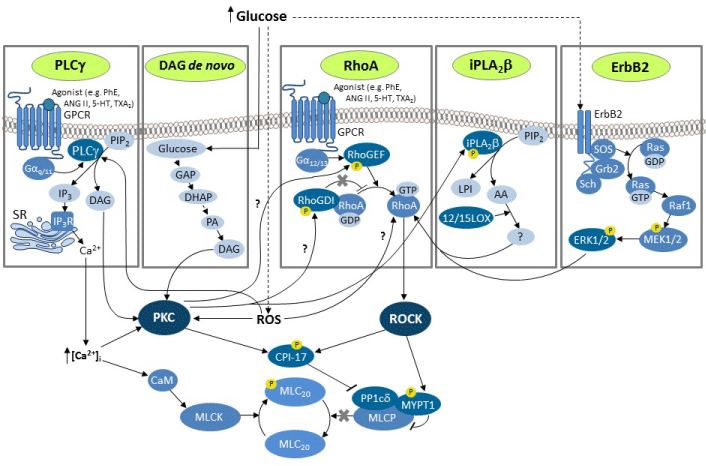
Figure 1: A scheme illustrating the PKC- and ROCK-mediated mechanisms of contractile proteins Ca2+ sensitization in vascular smooth muscle cells in DM. Hyperglycemia induces elevated ROS leading to PKC activation. Activated by hyperglycemia DAG de novo synthesis and ROS-mediated PLCγ-dependent DAG formation both also lead to PKC activation. Hyperglycemia increases DAG concentration by promptings de novo synthesis from the glycolytic intermediatesglyceraldehyde-3-phosphate (GAP) and dihydroxyacetonephosphate (DHAP), leading to the formation of phosphatidic acid (PA). High glucose, possibly mediated by ROS, and the ErbB2/ERK1/2-mediated pathway, also induces activation of RhoA and ROCK. Both PKC and ROCKactivate CPI-17 which binds to MLCP and inhibits its activity. ERK1/2 activation is mediated by the mitogen-activated protein kinases pathway involving Src homology 2 domain containing transforming protein (Sch), growth factor receptor-bound protein 2 (Grb-2), son of sevenless homologprotein (SOS),rat sarcoma protein (Ras), rapidly accelerated fibrosarcoma proto-oncogene protein kinase (Raf1), and mitogen-activated protein kinase kinases 1 and 2 (MEK1/2).The inhibition of the MLCP results in increased MLC20 phosphorylation for any given level of [Ca2+]i and activity of MLCK. High glucose-induced upregulation of PKC leads to activation of the iPLA2β/12/15- LOX pathway and upregulation of RhoA/ROCK. Also, PKC may mediate a high glucose-induced activation of RhoA/ROCKvia phosphorylation of RhoGDI or RhoGEF.
ROCK
The Rho-kinases or ROCKs is a small group of serine/threonine kinases, regulatory enzymes that also play an important role in vascular function regulation [39,40]. ROCKs are represented with two isoforms encoded by two different genes, ROCK-1 (ROCK I, P160-ROCK, or ROK β) and ROCK-2 (ROCK II or ROK α) [41-43]. Both isoforms are highly homologous and are expressed in vascular tissues [44-46]. ROCK is mainly dispersed in the cytoplasm, but is partially translocated to the peripheral membrane upon activation [42,47].
ROCK is activated by monomeric G proteins (small GTPases) of the Rho (Ras homolog) protein family [39,42,48-50]. The Rho protein family consists of at least 20 members classified into 5 groups: the Rho-like, Rac-like, Cdc42-like, Rnd, and RhoBTB subfamilies, whereas RhoD, Rif, and RhoH/TTF do not fall into any obvious grouping [50]. Proteins of the Rho family regulate a wide range of fundamental cell functions such as contraction, motility, proliferation, and apoptosis [50-52]. RhoA, belonging to the Rho-like subfamily, is the main upstream activator of ROCK [50]. Binding of the active GTP-bound form of RhoA to the Rho-binding domain stimulates the phosphotransferase activity of ROCK by disrupting the interaction between the catalytic and the inhibitory C-terminal regions of the enzyme [49]. However, the stimulatory effect of RhoA on ROCK activity is limited [53]. RhoA functions as a molecular switch which cycles between an inactive GDP-bound and active GTP-bound conformations. Activated RhoA interacts with downstream targets leading to cellular responses. Upon activation RhoA is translocated from the cytosol to the cell membrane [54,55]. Activation of Rho GTPases is regulated by several mechanisms including the activation of heterotrimeric G protein-coupled receptors [49]. Three types of regulators of Rho proteins have been identified. Rho guanine-nucleotide exchange factors (RhoGEF) promote the formation of the GTP-bound form of Rho proteins [50], whereas Rho GTPase activating protein (RhoGAP) accelerates the intrinsic GTPase activity resulting in the GDP-bound form [56]. Rho guanine nucleotide dissociation inhibitor (RhoGDI) binds to a subset of Rho proteins, inhibits nucleotide exchange and sequesters these proteins away from the membrane, where normally they would be active [50]. On the other hand, it has been shown that ROCK-1 activity can be inhibited by the Rho protein Rnd3/RhoE which binds to the N-terminal region of ROCK-1 and prevents RhoA binding to the Rho-binding domain [57]. Two other small G proteins of the Ras superfamily, Gem and Rad, have been shown to inhibit the ROCK function [58,59].
The Rho/ROCK signaling pathway has been implicated in the pathogenesis of diabetes. It has been demonstrated that ROCK activity is enhanced in vascular tissues in diabetes [31,44,48,60-63]. Recent publications demonstrated that diabetes type 1-induced vascular dysfunction in the rat aorta arises due to an upregulation of ROCK- 2 isoform [64]. An increased level of ROCK-2 protein expression in corpus cavernosum of diabetic mice [65] and retinal vessels of type 1 model diabetic rats [66] has also been reported. ROCK-1 gene and protein upregulation has also been reported from different vessels in various diabetic animal models [61,62,67]. The mRNA and protein levels of RhoA is also increased in arteries of diabetic rats and mice [48,62,65,68-70,71]. The mechanism of RhoA and ROCK activation by high glucose levels remains to be defined. However ROS, thought to play a central role in the pathogenesis of vascular diabetic complications, have been shown to activate RhoA [72-74, 75]. In another study, the authors reported that glucose-induced calcium-independent phospholipase A2β (iPLA2β) upregulation may activate the RhoA/ROCK via 12/15-lipoxygenases in vascular SMCs [76]. On the other hand, it has been suggested that activation of ROCK in DM and high glucose level can be mediated by ErbB2 and Ras/ Raf/extracellular signal-regulated kinase 1/2 (ERK1/2) [77]. ErbB2 is a transmembrane tyrosine kinase receptor of the epidermal growth factor receptor (ErbB/EGFR) family which downstream effector in vascular SMCs is ERK1/2 [77] (Figure).
PKC-mediated SMCs Ca2+ sensitization in DM
While numerous studies have demonstrated that DM impairs vascular function by inhibiting endothelium-dependent vasodilatation [9,11-14], others have shown that DM enhances vascular contractility in an endothelium-independent manner [31,38,63,78-80]. A large number of studies suggest that vascular smooth muscle contractile responses are dramatically enhanced in diabetes [31,48,78,80-84]. Studies on different diabetic animal models have shown that α1- adrenomimetics-, serotonin-(or 5-hydroxytryptamine, 5-HT), and angiotensin II-induced SMC contraction is enhanced in arteries [31,37,48,67,77,79,81,84-88], and PKC-α,-ε, and �δ isozimes contribute to this process [31,48,63,76,78]. Similarly, endothelium-independent vasoconstriction mediated by prostaglandin E receptor 1 (EP1) and 3 (EP3) activation has been shown to be enhanced in type 2 diabetic rats’ mesenteric arteries and highly sensitive to PKC-δ inhibition [83].
It is well known that vascular smooth muscle contractile responses are primarily triggered by increased intracellular concentration of Ca2+ ([Ca2+]i). Both increased Ca2+ influx and release of Ca2+ from intracellular stores have been proposed to be involved in DM, contributing to the enhanced vascular reactivity [85,89]. There is evidence that stimulation of arteries with α1- adrenomimetics is associated with increased Ca2+ influx in diabetic vessels over normal ones [82,85,90,91]. Other authors have also reported increased intracellular Ca2+ to be involved in the diabetic vascular hyperreactivity [82,89]. However, there is a controversy here with reports of inhibition of voltage- and store-operated Ca2+ entry channels in vascular SMC in DM [92,93]. Alternatively, Ca2+ sensitization of the contractile proteins has been proposed as a more general mechanism of the elevated DM-associated vascular contractility [48,68,78,80]. One striking observation supporting this view is the enhanced contractile response to α1-adrenomimetics by arteries from diabetic rats and mice which is not associated with [Ca2+]I changes [31,86]. Similarly, under high glucose conditions, thromboxane A2-induced aortic smooth muscle contraction has also been shown to increase independently from intracellular calcium concentration [37].
Phosphorylation and dephosphorylation of the 20-kDa myosin light chain (MLC) are the major regulatory mechanisms of smooth muscle contractility. Increased [Ca2+]I activates calmodulin-dependent MLC kinase (MLCK) which catalyzes the phosphorylation of MLC at Thr18 and Ser19 leading to actin-myosin interaction and vascular smooth muscles contraction [94,95]. On top of this primary regulatory pathway, several modulatory pathways exist in smooth muscles that can alter the magnitude of the force that is developed at any given [Ca2+]I [94,95]. Both the PKC-mediated [96,97] and ROCK-mediated [39,49,97] mechanisms have been shown to be involved in elevated myofilament sensitivity to Ca2+.
These two pathways converge to phosphorylate an inhibitory protein CPI-17 at Thr38 [95,96,98,99] (see Figure). The CPI-17 is the smooth muscle myosin light chain phosphatase (MLCP) PKC-dependent protein phosphatase-1 (PP1) inhibitor of 17 kDa [95,96,98,99]. The MLCP holoenzyme of smooth muscles is a heterotrimer consisting of three subunits: the130-kDa, 38-kDa, and 21-kDa subunit [100-102]. The 38-kDa subunit is identified as PKC-dependent protein phosphatase-1 (PP1) catalytic subunit (PP1cδ). The 130-kDa subunit is a regulatory and myosin-bound MLCP targeting subunit 1 (MYPT1) [100-103]. The 21-kDa subunit’s function of MLCP is unclear [104]. Phosphorylated CPI-17 directly binds to MLCP and inhibits its activity [105]. Inhibition of MLCP results in a higher level of MLC phosphorylation for any given level of [Ca2+]i and activity of MLCK [106]. This phenomenon, known as Ca2+ sensitization of smooth muscles, could thereby maintain vascular contraction [49,94].
Mueed and coauthors have reported that enhanced contractile responses of mesenteric arteries from streptozotocin (STZ)-induced diabetic rats upon stimulation of α1-adrenoceptors are accompanied by elevated levels of PKC-α and -ε, and PKC-dependent elevation in CPI-17 phosphorylation in vascular SMCs [77]. We have recently shown that elevated vascular SMC contractility in STZ-diabetic rats is mediated by Ca2+ sensitization, and PKC is contributing to this process [80]. It has also been shown recently that PKC-δ mediates Ca2+ sensitization in the intrarenal interlobar artery of type 2 diabetic ob/ob mice [31].
ROCK-mediated SMCs Ca2+ sensitization in DM
Numerous studies have demonstrated that enhanced contractile responses to α-adrenomimetics, angiotensin II, and 5-THin vessels from animal models of both types of DM [13,31,48,67,77,82,84,107] as well as in pre-diabetic obese Zucker rats [108] are mediated by ROCK.
For increase in myofilament Ca2+ sensitivity in the DMROCK-mediated mechanism have been shown [39,48,49,80,98,103,108,109]. In this mechanism, ROCK phosphorylates CPI-17 at Thr38 [95,96,98,99,110-114] which then directly binds to PP1cδ and inhibits the activity of MLCP [105, 115-117] (see Figure). In addition, ROCK directly phosphorylates the MYPT1 subunit of MLCP at Thr855 and/or Thr850, Thr695, Thr696, and Thr697 [39,49,98,103,106,109,114,118- 121] that also consequently inhibits the phosphatase activity.
In smooth muscle cells from STZ-induced model of type 1 diabetes rats, phosphorylation levels of MYPT1 in aorta has been shown to be significantly elevated [64]. Similar results have been obtained from the saphenous vein of patients with DM by Matsuo and coauthors, which have shown that the hyper reactivity to 5-HT in smooth muscles of these vessels is due to higher phosphorylation of MLCP, evident from elevation of the P(Thr696)-MYPT1 to total MYPT1 ratio [121].
Our research group has shown that elevated vascular SMC contractility in rats with STZ-induced DM is associated with enhanced Ca2+ sensitivity of contractile myofilaments and both ROCK and PKC are clearly contribute to this process in diabetic vasculature [80]. ROCK-mediated calcium sensitization has been shown to be responsible for hypertension development in type 2 diabetes as well [31,68]. It has also been suggested recently that both PKC-δ and ROCK mediate Ca2+ sensitization in smooth muscles of the intrarenal interlobar artery of type 2 diabetic (ob/ob) mice [31]. It has been shown that a1-adrenoceptor-mediated vasoconstriction in penile [82] and gracilis arteries [107] from rat models of prediabetes/ metabolic syndrome (obese Zucker rats) also involves ROCK-dependent Ca2+ sensitization. Other authors have demonstrated that ROCK-mediated CPI-17 phosphorylation increase in vascular SMCs of the type 2 diabetic db/db mice model and SMCs cultured in presence of high glucose concentration [48]. Both activation of the ROCK pathways that phosphorylates CPI-17 and increase in total CPI-17 protein level seem to be involved [48], and CPI-17 up-regulation/activation in type 2 diabetic db/db mice vasculature is associated with a significant blood pressure increase [122].
A number of papers have reported a link between the ROCK and PKC pathways in diabetes or under high glucose conditions. Some of them suggest that ROCK is upstream of PKC in diabetic bladder [123] and diabetic cardiomyocytes [124], whereas in diabetic vascular SMC ROCK has been reported to be downstream of PKC [48,77]. Xie and coauthors have shown that PKC in vascular SMC is required for high glucose-induced ROCK activation and consequently CPI-17 phosphorylation [48]. These authors also suggest that, although PKC can directly phosphorylate CPI-17 upon some agonist stimulation in the vascular smooth muscle tissue under physiological conditions, PKC is not the kinase that directly phosphorylates CPI-17 in the presence of high glucose [48]. It has been shown that ROCK mRNA and protein levels can be upregulated through PKC and it is luckily that under diabetic conditions RhoA/ROCK and CPI-17 are downstream effectors of PKC [48]. More recently, the same group of researchers has shown that high glucose-induced activation of PKC leading to activation of iPLA2β and up-regulation of RhoA/ROCK via 12/15-lipoxygenases, thereby contributes to diabetes-associated vascular smooth muscle Ca2+ sensitization and hypercontractility [76]. Products of the phospholipase A2 enzymes group include arachidonic acid which can be rapidly metabolized to a variety of mediators by lipoxygenases and other oxygenases to a number of bioactive eicosanoids [125]. However, it remains unclear how the catalytic activity of the 12/15-lipoxygenase may activate RhoA/ ROCK/CPI-17.It is possible to assume, that PKCmay mediate high glucose-induced activation of RhoA via phosphorylation of RhoGDI [126] or RhoGEF [127] (Figure).
Conclusion
The presented data suggest that PKC and ROCK in vascular SMC highly contribute to enhanced vascular tone and arterial hypercontractility in diabetes by mediating SMC Ca2+-sensitization. Our review shows that PKC and ROCK are potential therapeutic targets for treating vascular diabetic complications. Developing innovative pharmacological approaches that could modulate PKC and ROCK activity is highly important for new strategies that might prove clinical relevancy in preventing the development and/or retarding the progression of diabetes-associated vascular complications.
References
- Unwin N, Gan D, Whiting D. The IDF Diabetes Atlas: providing evidence, raising awareness and promoting action. Diabetes Res Clin Pract. 2010; 87: 2-3.
- Zimmet PZ. The growing pandemic of type 2 diabetes: a crucial need for prevention and improved detection. Medicographia. 2011; 33: 15-21.
- Madonna R, De Caterina R. Cellular and molecular mechanisms of vascular injury in diabetes--part I: pathways of vascular disease in diabetes. Vascul Pharmacol. 2011; 54: 68-74.
- Whiteley L, Padmanabhan S, Hole D, Isles C. Should diabetes be considered a coronary heart disease risk equivalent? results from 25 years of follow-up in the Renfrew and Paisley survey. Diabetes Care. 2005; 28: 1588-1593.
- Funk SD, Yurdagul A Jr, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. Int J Vasc Med. 2012; 2012: 569654.
- Arora MK, Singh UK. Molecular mechanisms in the pathogenesis of diabetic nephropathy: an update. Vascul Pharmacol. 2013; 58: 259-271.
- Arora MK, Singh UK. Molecular mechanisms in the pathogenesis of diabetic nephropathy: an update. Vascul Pharmacol. 2013; 58: 259-271.
- Behnam-Rassouli M, Ghayour MB, Ghayour N. Microvascular complications of diabetes. J Biol Sci. 2010; 10: 411-423.
- Pitocco D, Zaccardi F, Di Stasio E, Romitelli F, Santini SA, Zuppi C, et al. Oxidative stress, nitric oxide, and diabetes. Rev Diabet Stud. 2010; 7: 15-25.
- Potenza MA, Nacci C, Gagliardi S, Montagnani M. Cardiovascular complications in diabetes: lessons from animal models. Curr Med Chem. 2011; 18: 1806-1819.
- Naka KK, Papathanassiou K, Bechlioulis A, Kazakos N, Pappas K, Tigas S, et al. Determinants of vascular function in patients with type 2 diabetes. Cardiovasc Diabetol. 2012; 11: 127.
- Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med. 2012; 2012: 918267.
- Pearson JT, Jenkins MJ, Edgley AJ, Sonobe T, Joshi M, Waddingham MT. Acute Rho-kinase inhibition improves coronary dysfunction in vivo, in the early diabetic microcirculation. Cardiovasc Diabetol. 2013; 12: 111.
- Klymenko K, Novokhatska T, Kizub I, Parshikov A, Dosenko V, Soloviev A, et al. PKC-δ isozyme gene silencing restores vascular function in diabetic rat. J Basic Clin Physiol Pharmacol. 2014.
- Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, Lafont S, Bergeonneau C, Kassai B, et al. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. BMJ. 2011; 343: d4169.
- Sedeek M, Montezano AC, Hebert RL, Gray SP, Di Marco E, Jha JC. Oxidative stress, Nox isoforms and complications of diabetes--potential targets for novel therapies. J Cardiovasc Transl Res. 2012; 5: 509-518.
- Schaffer SW, Jong CJ, Mozaffari M. Role of oxidative stress in diabetes-mediated vascular dysfunction: unifying hypothesis of diabetes revisited. Vascul Pharmacol. 2012; 57: 139-149.
- Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010; 106: 1319-1331.
- Clarke M, Dodson PM. PKC inhibition and diabetic microvascular complications. Best Pract Res Clin Endocrinol Metab. 2007; 21: 573-586.
- Kizub IV, Klymenko KI, Soloviev AI. Protein kinase C in enhanced vascular tone in diabetes mellitus. Int J Cardiol. 2014; 174: 230-242.
- Cosentino-Gomes D, Rocco-Machado N, Meyer-Fernandes JR. Cell Signaling through Protein Kinase C Oxidation and Activation. Int J Mol Sci. 2012; 13: 10697-10721.
- Steinberg S. Structural basis of protein kinase C isoform function. Physiol Rev. 2008; 88: 1341-1378.
- Sobhia ME, Grewal BK, Bhat J, Rohit S, Punia V. Protein kinase C bII in diabetic complications: survey of structural, biological and computational studies. Expert Opin Ther Targets. 2012; 16: 325-344.
- Cenni V, Döppler H, Sonnenburg ED, Maraldi N, Newton AC, Toker A, et al. Regulation of novel protein kinase C epsilon by phosphorylation. Biochem J. 2002; 363: 537-545.
- Duquesnes N, Lezoualc'h F, Crozatier B. PKC-delta and PKC-epsilon: foes of the same family or strangers? J Mol Cell Cardiol. 2011; 51: 665-673.
- Wang QJ. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol Sci. 2006; 27: 317-323.
- Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl AcadSci USA. 1992; 89: 11059-11063.
- Kang N, Alexander G, Park JK, Maasch C, Buchwalow I, Luft FC, et al. Differential expression of protein kinase C isoforms in streptozotocin-induced diabetic rats. Kidney Int. 1999; 56: 1737-1750.
- Tickerhoof MM, Farrell PA, Korzick DH. Alterations in rat coronary vasoreactivity and vascular protein kinase C isoforms in Type 1 diabetes. Am J Physiol Heart Circ Physiol. 2003; 285: H2694-2703.
- Ramana KV, Friedrich B, Tammali R, West MB, Bhatnagar A, Srivastava SK. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes. 2005; 54: 818-829.
- Nobe K, Takenouchi Y, Kasono K, Hashimoto T, Honda K. Two Types of Overcontraction Are Involved in Intrarenal Artery Dysfunction in Type II Diabetic Mouse. J Pharmacol Exp Ther. 2014; 351: 77-86.
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010; 107: 1058-1070.
- Son SM. Reactive oxygen and nitrogen species in pathogenesis of vascular complications of diabetes. Diabetes Metab J. 2012; 36: 190-198.
- Inoguchi T, Sonta T, Tsubouchi H, Etoh T, Kakimoto M, Sonoda N, et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol. 2003; 14: S227-232.
- Gao L, Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res. 2009; 82: 9-20.
- Du X, Matsumura T, Edelstein D, Rossetti L, Zsengellér Z, Szabó C. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003; 112: 1049-1057.
- Nobe K, Suzuki H, Nobe H, Sakai Y, Momose K. High-glucose enhances a thromboxane A2-induced aortic contraction mediated by an alteration of phosphatidylinositol turnover. J Pharmacol Sci. 2003; 92: 267-282.
- Nobe K, Miyatake M, Nobe H, Sakai Y, Takashima J, Momose K. Novel diacylglycerol kinase inhibitor selectively suppressed an U46619-induced enhancement of mouse portal vein contraction under high glucose conditions. Brit J Pharmacol. 2004; 143: 166-178.
- Schofield AV, Bernard O. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit Rev Biochem Mol Biol. 2013; 48: 301-316.
- Grisk O. Potential benefits of rho-kinase inhibition in arterial hypertension. Curr Hypertens Rep. 2013; 15: 506-513.
- Leung T, Manser E, Tan L, Lim L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem. 1995; 270: 29051-29054.
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996; 15: 2208-2216.
- Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996; 392: 189-193.
- Nuno DW, Harrod JS, Lamping KG. Sex-dependent differences in Rho activation contribute to contractile dysfunction in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2009; 297: H1469-1477.
- Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, et al. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res. 2009; 104: 531-540.
- Rees RW, Ziessen T, Ralph DJ, Kell P, Moncada S, Cellek S. Human and rabbit cavernosal smooth muscle cells express Rho-kinase. Int J Impot Res. 2002; 14: 1-7.
- Leung T, Manser E, Tan L, Lim L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem. 1995; 270: 29051-29054.
- Xie Z, Su W, Guo Z, Pang H, Post SR, Gong MC. Up-regulation of CPI-17 phosphorylation in diabetic vasculature and high glucose cultured vascular smooth muscle cells. Cardiovasc Res. 2006; 69: 491-501.
- Loirand G, Guérin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006; 98: 322-334.
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004; 116: 167-179.
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002; 420: 629-635.
- Ishikura S, Koshkina A, Klip A. Small G proteins in insulin action: Rab and Rho families at the crossroads of signal transduction and GLUT4 vesicle traffic. Acta Physiol (Oxf). 2008; 192: 61-74.
- Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N. Rho-associated kinase of chicken gizzard smooth muscle. J Biol Chem. 1999; 274: 3744-3752.
- Takaishi K, Sasaki T, Kameyama T, Tsukita S, Tsukita S, Takai Y, et al. Translocation of activated Rho from the cytoplasm to membrane ruffling area, cell-cell adhesion sites and cleavage furrows. Oncogene. 1995; 11: 39-48.
- Gong MC, Fujihara H, Somlyo AV, Somlyo AP. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J Biol Chem. 1997; 272: 10704-10709.
- Bernards A, Settleman J. GAP control: regulating the regulators of small GTPases. Trends Cell Biol. 2004; 14: 377-385.
- Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003; 23: 4219-4229.
- Ward Y, Yap SF, Ravichandran V, Matsumura F, Ito M, Spinelli B. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J Cell Biol. 2002; 157: 291-302.
- Chardin P. GTPase regulation: getting aRnd Rock and Rho inhibition. Curr Biol. 2003; 13: R702-704.
- Begum N, Duddy N, Sandu O, Reinzie J, Ragolia L. Regulation of myosin-bound protein phosphatase by insulin in vascular smooth muscle cells: evaluation of the role of Rho kinase and phosphatidylinositol-3-kinase-dependent signaling pathways. Mol Endocrinol. 2000; 14: 1365-1376.
- Chang S, Hypolite JA, Changolkar A, Wein AJ, Chacko S, DiSanto ME. Increased contractility of diabetic rabbit corpora smooth muscle in response to endothelin is mediated via Rho-kinase beta. Int J Impot Res. 2003; 15: 53-62.
- Bivalacqua TJ, Champion HC, Usta MF, Cellek S, Chitaley K, Webb RC, et al. RhoA/Rho-kinase suppresses endothelial nitric oxide synthase in the penis: a mechanism for diabetes-associated erectile dysfunction. Proc Natl Acad Sci U S A. 2004; 101: 9121-9126.
- Akhtar S, Yousif MH, Dhaunsi GS, Sarkhouh F, Chandrasekhar B, Attur S. Activation of ErbB2 and Downstream Signalling via Rho Kinases and ERK1/2 Contributes to Diabetes-Induced Vascular Dysfunction. PLoS One. 2013; 8: e67813.
- Cicek FA, Kandilci HB, Turan B. Role of ROCK upregulation in endothelial and smooth muscle vascular functions in diabetic rat aorta. Cardiovasc Diabetol. 2013; 12: 51.
- Toque HA, Nunes KP, Yao L, Liao JK, Webb RC, Caldwell RB, et al. Activated Rho kinase mediates diabetes-induced elevation of vascular arginase activation and contributes to impaired corpora cavernosa relaxation: Possible involvement of p38 MAPK activation. J Sex Med. 2013; 10: 1502-1515.
- Arita R, Hata Y, Nakao S, Kita T, Miura M, Kawahara S. Rho kinase inhibition by fasudil ameliorates diabetes-induced microvascular damage. Diabetes. 2009; 58: 215-226.
- Failli P, Alfarano C, Franchi-Micheli S, Mannucci E, Cerbai E, Mugelli A. Losartan counteracts the hyper-reactivity to angiotensin II and ROCK1 over-activation in aortas isolated from streptozotocin-injected diabetic rats. Cardiovasc Diabetol. 2009; 8: 32.
- Sandu OA, Ragolia L, Begum N. Diabetes in the Goto-Kakizaki rat is accompanied by impaired insulin-mediated myosin-bound phosphatase activation and vascular smooth muscle cell relaxation. Diabetes. 2000; 49: 2178-2189.
- Miao L, Calvert JW, Tang J, Zhang JH. Upregulation of small GTPase RhoA in the basilar artery from diabetic (mellitus) rats. Life Sci. 2002; 71: 1175-1185.
- Massey AR, Miao L, Smith BN, Liu J, Kusaka I, Zhang JH. Increased RhoA translocation in renal cortex of diabetic rats. Life Sci. 2003; 72: 2943-2952.
- El-Remessy AB, Tawfik HE, Matragoon S, Pillai B, Caldwell RB, Caldwell RW, et al. Peroxynitrite mediates diabetes-induced endothelial dysfunction: possible role of Rho kinase activation. Exp Diabetes Res. 2010; 2010: 247861.
- Whiteside CI. Cellular mechanisms and treatment of diabetes vascular complications converge on reactive oxygen species. Curr Hypertens Rep. 2005; 7: 148-154.
- Bailey SR, Mitra S, Flavahan S, Flavahan NA. Reactive oxygen species from smooth muscle mitochondria initiate cold-induced constriction of cutaneous arteries. Am J Physiol Heart Circ Physiol. 2005; 289: H243-250.
- Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007; 21: 3666-3676.
- Knock GA, Ward JP. Redox regulation of protein kinases as a modulator of vascular function. Antioxid Redox Signal. 2011; 15: 1531-1547.
- Xie Z, Gong MC, Su W, Xie D, Turk J, Guo Z. Role of calcium-independent phospholipase A2beta in high glucose-induced activation of RhoA, Rho kinase, and CPI-17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals. J Biol Chem. 2010; 285: 8628-8638.
- Akhtar S, Yousif MH, Dhaunsi GS, Sarkhouh F, Chandrasekhar B, Attur S, et al. Activation of ErbB2 and Downstream Signalling via Rho Kinases and ERK1/2 Contributes to Diabetes-Induced Vascular Dysfunction. PLoS One. 2013; 8: e67813.
- Mueed I, Zhang L, MacLeod KM. Role of the PKC/CPI-17 pathway in enhanced contractile responses of mesenteric arteries from diabetic rats to alpha-adrenoceptor stimulation. Br J Pharmacol. 2005; 146: 972-982.
- Schulingkamp RJ, Aloyo V, Tallarida RJ, Raffa RB. Changes in aorta alpha1-adrenoceptor number and affinity during one year of streptozotocin-induced diabetes in rats. Pharmacology. 2005; 74: 23-30.
- Kizub IV, Pavlova OO, Johnson CD, Soloviev AI, Zholos AV. Rho kinase and protein kinase C involvement in vascular smooth muscle myofilament calcium sensitization in arteries from diabetic rats. Br J Pharmacol. 2010; 159: 1724-1731.
- Lee J-H, Bahk J-H, Park S-H, Huh J. The diabetes-induced functional and distributional changes of the alpha 1-adrenoceptor of the abdominal aorta and distal mesenteric artery from streptozotocin-induced diabetic rats. Korean J Anesthesiol. 2011; 60: 272-281.
- Villalba N, Contreras C, Hernández M, García-Sacristán A, Prieto D. Impaired Ca2+ handling in penile arteries from prediabetic Zucker rats: involvement of Rho kinase. Am J Physiol Heart Circ Physiol. 2011; 300: H2044-2053.
- Ishida K, Matsumoto T, Taguchi K, Kamata K, Kobayashi T. Protein kinase C delta contributes to increase in EP3 agonist-induced contraction in mesenteric arteries from type 2 diabetic Goto-Kakizaki rats. Pflugers Arch. 2012; 463: 593-602.
- Matsumoto T, Watanabe S, Taguchi K, Kobayashi T. Mechanisms underlying increased serotonin-induced contraction in carotid arteries from chronic type 2 diabetic Goto-Kakizaki rats. Pharmacol Res. 2014; 87: 123-132.
- White RE, Carrier GO. Vascular contraction induced by activation of membrane calcium ion channels is enhanced in streptozotocin-diabetes. J Pharmacol Exp Ther. 1990; 253: 1057-1062.
- Chow WL, Zhang L, MacLeod KM. Noradrenaline-induced changes in intracellular Ca(2+) and tension in mesenteric arteries from diabetic rats. Br J Pharmacol. 2001; 134: 179-187.
- Nobe K, Sakai Y, Nobe H, Momose K. Dysfunction of aorta involves different patterns of intracellular signaling pathways in diabetic rats. Eur J Pharmacol. 2003; 471: 195-204.
- Okon EB, Szado T, Laher I, McManus B, van Breemen C. Augmented contractile response of vascular smooth muscle in a diabetic mouse model. J Vasc Res. 2003; 40: 520-530.
- Balasubramanyam M, Balaji RA, Subashini B, Mohan V. Evidence for mechanistic alterations of Ca2+ homeostasis in Type 2 diabetes mellitus. Int J Exp Diabetes Res. 2001; 1: 275-287.
- Mahmoudian M, Behnaz F, Rezaei E. Diabetes-induced changes in the contractility of the aorta and pA2 of nifedipine in the rat. Acta Diabetol. 1996; 33: 114-117.
- Ungvari Z, Pacher P, Kecskemeti V, Papp G, Szollár L, Koller A. Increased myogenic tone in skeletal muscle arterioles of diabetic rats. Possible role of increased activity of smooth muscle Ca2+ channels and protein kinase C. Cardiovasc Res. 1999; 43: 1018-1028.
- Wang R, Wu Y, Tang G, Wu L, Hanna ST. Altered L-type Ca(2+) channel currents in vascular smooth muscle cells from experimental diabetic rats. Am J Physiol Heart Circ Physiol. 2000; 278: H714-722.
- Curtis TM, Major EH, Trimble ER, Scholfield CN. Diabetes-induced activation of protein kinase C inhibits store-operated Ca2+ uptake in rat retinal microvascular smooth muscle. Diabetologia. 2003; 46: 1252-1259.
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003; 83: 1325-1358.
- Schubert R, Lidington D, Bolz SS. The emerging role of Ca2+ sensitivity regulation in promoting myogenic vasoconstriction. Cardiovasc Res. 2008; 77: 8-18.
- Senba S, Eto M, Yazawa M. Identification of trimeric myosin phosphatase (PP1M) as a target for a novel PKC-potentiated protein phosphatase-1 inhibitory protein (CPI17) in porcine aorta smooth muscle. J Biochem. 1999; 125: 354-362.
- Salamanca DA, Khalil RA. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol. 2005; 70: 1537-1547.
- Swärd K, Mita M, Wilson DP, Deng JT, Susnjar M, Walsh MP, et al. The role of RhoA and Rho-associated kinase in vascular smooth muscle contraction. Curr Hypertens Rep. 2003; 5: 66-72.
- Niiro N, Koga Y, Ikebe M. Agonist-induced changes in the phosphorylation of the myosin- binding subunit of myosin light chain phosphatase and CPI17, two regulatory factors of myosin light chain phosphatase, in smooth muscle. Biochem J. 2003; 369: 117-128.
- Shimizu H, Ito M, Miyahara M, Ichikawa K, Okubo S, Konishi T. Characterization of the myosin-binding subunit of smooth muscle myosin phosphatase. J Biol Chem. 1994; 269: 30407-30411.
- Okubo S, Ito M, Takashiba Y, Ichikawa K, Miyahara M, Shimizu H, et al. A regulatory subunit of smooth muscle myosin bound phosphatase. Biochem Biophys Res Commun. 1994; 200: 429-434.
- Ito M, Nakano T, Erdodi F, Hartshorne DJ. Myosin phosphatase: structure, regulation and function. Mol Cell Biochem. 2004; 259: 197-209.
- Ichikawa K, Ito M, Hartshorne DJ. Phosphorylation of the large subunit of myosin phosphatase and inhibition of phosphatase activity. J Biol Chem. 1996; 271: 4733-4740.
- Takeya K, Wang X, Sutherland C, Kathol I, Loutzenhiser K, Loutzenhiser RD, et al. The involvement of myosin regulatory light chain diphosphorylation in sustained vasoconstriction under pathophysiological conditions J. Smooth Muscle Res. 2014; 50: 18-28.
- Eto M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J Biol Chem. 2009; 284: 35273-35277.
- Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999; 274: 37385-37390.
- Didion SP, Lynch CM, Faraci FM. Cerebral vascular dysfunction in TallyHo mice: a new model of Type II diabetes. Am J Physiol Heart Circ Physiol. 2007; 292: H1579-1583.
- Naik JS, Xiang L, Hester RL. Enhanced role for RhoA-associated kinase in adrenergic-mediated vasoconstriction in gracilis arteries from obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2006; 290: R154-161.
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996; 273: 245-248.
- Eto M, Ohmori T, Suzuki M, Furuya K, Morita F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J Biochem. 1995; 118: 1104-1107.
- Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, et al. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000; 475: 197-200.
- Hayashi Y, Senba S, Yazawa M, Brautigan DL, Eto M. Defining the structural determinants and a potential mechanism for inhibition of myosin phosphatase by the protein kinase C-potentiated inhibitor protein of 17 kDa. J Biol Chem. 2001; 276: 39858-39863.
- Ohki S, Eto M, Kariya E, Hayano T, Hayashi Y, Yazawa M, et al. Structure of the myosin phosphatase inhibitor protein CPI-17 shows phosphorylation-induced conformational changes responsible for activation. J Mol Biol. 2001; 314: 839-849.
- Pang H, Guo Z, Su W, Xie Z, Eto M, Gong MC. RhoA-Rho kinase pathway mediates thrombin- and U-46619-induced phosphorylation of a myosin phosphatase inhibitor, CPI-17, in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2005; 289: C352-360.
- Eto M, Senba S, Morita F, Yazawa M. Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI-17) in smooth muscle: its specific localization in smooth muscle. FEBS Lett. 1997; 410: 356-360.
- Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem. 2000; 275: 9897-9900.
- Dubois T, Howell S, Zemlickova E, Learmonth M, Cronshaw A, Aitken A, et al. Novel in vitro and in vivo phosphorylation sites on protein phosphatase 1 inhibitor CPI-17. Biochem Biophys Res Commun. 2003; 302: 186-192.
- Velasco G, Armstrong C, Morrice N, Frame S, Cohen P. Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1M at Thr850 induces its dissociation from myosin. FEBS Lett. 2002; 527: 101-104.
- Murányi A, Derkach D, Erdodi F, Kiss A, Ito M, Hartshorne DJ. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett. 2005; 579: 6611-6615.
- Wilson DP, Susnjar M, Kiss E, Sutherland C, Walsh MP. Thromboxane A2-induced contraction of rat caudal arterial smooth muscle involves activation of Ca2+ entry and Ca2+ sensitization: Rho-associated kinase-mediated phosphorylation of MYPT1 at Thr-855, but not Thr-697. Biochem J. 2005; 389: 763-774.
- Matsuo Y, Kuwabara M, Tanaka-Totoribe N, Kanai T, Nakamura E, Gamoh S, et al. The defective protein level of myosin light chain phosphatase (MLCP) in the isolated saphenous vein, as a vascular conduit in coronary artery bypass grafting (CABG), harvested from patients with diabetes mellitus (DM). Biochem Biophys Res Commun. 2011; 412: 323-327.
- Su W, Guo Z, Randall DC, Cassis L, Brown DR, Gong MC. Hypertension and disrupted blood pressure circadian rhythm in type 2 diabetic db/db mice. Am J Physiol Heart Circ Physiol. 2008; 295: H1634-1641.
- Nobe K, Yamazaki T, Tsumita N, Hashimoto T, Honda K. Glucose-dependent enhancement of diabetic bladder contraction is associated with a rho kinase-regulated protein kinase C pathway. J Pharmacol Exp Ther. 2009; 328: 940-950.
- Soliman H, Gador A, Lu YH, Lin G, Bankar G, MacLeod KM, et al. Diabetes-induced increased oxidative stress in cardiomyocytes is sustained by a positive feedback loop involving Rho kinase and PKCβ2. Am J Physiol Heart Circ Physiol. 2012; 303: H989-989H1000.
- Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994; 269: 13057-13060.
- Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem. 2001; 276: 22614-22620.
- Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase Calpha-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem. 2003; 278: 28793-28798.